

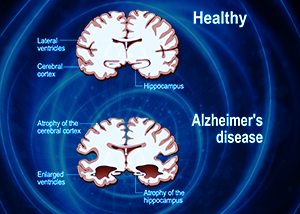
As humans, we can all be guaranteed of one similar destiny: we will all get old and we will all eventually die. Nonetheless, nothing strikes fear in the hearts of a human being more than the prospect of dying a slow, uncomfortable death. One of the worst such types of slow decline is known by the name of dementia, and it is the loss of the very faculties that make us human. In dementia, every single faculty of the mind is affected, including memory, spatial and geographical orientation, comprehension, the ability to learn, language, and even judgment.
What is so insidious about the disease is that while all of these capabilities are gradually fading and coming apart, consciousness is undisturbed. The sufferer must watch silently as the body and mind gradually disintegrate, along with emotions, behavior, and the basic will to live. This gradual, deeply humiliating onset of symptoms is thought to begin in the brain and is described by a wide variety of neurological pathologies, the most common being Alzheimer’s disease.
All manifestations of this disease, in their deepest stages, lead to a complete disconnection from reality as we know it. In addition to the inability to recognize close friends or relatives, there is the additional insult of not knowing where or when one is. As dementia progresses to its final stages, a person is completely dependent on help from others, though everyone that is supporting the patient must suffer constant pressures and challenges due to the erratic behavior and, often, aggression that accompanies a complete loss of mind.
As such, dementia sufferers are often in deep conflict with not only themselves but also with those around them, and this makes providing health care for such individuals not only problematic but also extremely costly. Even the most patient healthcare providers can be provoked to react in the most challenging of times.
Alzheimer’s disease comprises approximately 60-70% of all dementia cases, but there are other forms as well, including vascular dementia, dementia with Lewy bodies, and dementia that is confined to the frontotemporal lobe of the brain. There is a wide variety of symptoms and manifestations of dementia, not all of which fit the classical description for Alzheimer’s, and it has historically been difficult to categorize many cases, due to complexity and overlap. Nonetheless, all forms of dementia have one thing in common: progressive, irreversible brain damage and a prolonged, uncomfortable end-of-life.
According to the World Health Organization, 50 million people worldwide currently have some form of this disease, 60% of which live in low- to middle-income countries. It is estimated that between 5 and 10% of the general population over 60, worldwide, has dementia. And this problem is not showing any signs of slowing down. Predictions indicate that by 2030, total dementia cases will reach 82 million, and the number is expected to almost double to 152 by 2050.
Many experts claim that the rising incidence of dementia is almost exclusively due to an increasing worldwide population combined with longer lifespan. Unfortunately, there are many risks hidden in our environment that are silently destroying our health from the bottom up without us knowing it. With no treatment currently in sight to effectively mitigate or reverse this disease, understanding its triggers and the process by which it develops is of paramount importance.1
The Growing MCT Epidemic
 Most scientists and doctors would argue that anything inducing or provoking oxidative stress in the brain has the potential to damage neurons and initiate the cascade that becomes dementia. I would not argue with them. Nonetheless, I have noticed a very alarming health trend over the last decade that involves an increasingly popular dietary choice – a choice that is silently creating problems of a magnitude that few suspect. I am talking about the now ubiquitous consumption of MCT (medium-chain triglyceride) oil and the havoc it is slowly but surely wreaking on the health of millions of people around the globe.
Most scientists and doctors would argue that anything inducing or provoking oxidative stress in the brain has the potential to damage neurons and initiate the cascade that becomes dementia. I would not argue with them. Nonetheless, I have noticed a very alarming health trend over the last decade that involves an increasingly popular dietary choice – a choice that is silently creating problems of a magnitude that few suspect. I am talking about the now ubiquitous consumption of MCT (medium-chain triglyceride) oil and the havoc it is slowly but surely wreaking on the health of millions of people around the globe.
Before you jump quickly to the conclusion that this is simply another article about the dangers of fats, please take some time to absorb the multiple points that will be carefully and thoroughly elaborated below. Numerous studies have been performed on the effects of medium chain triglycerides on the brain, the heart, the digestive tract, mitochondria, and immune system, but to date, a comprehensive and holistic view of its effects has not been posited to the degree that the average citizen can make an educated decision about its potential risks for health. Most of what is seen in blogs and social media is wholly positive, with only infrequent mentions of GI disturbances or allergic sensitization. In this article, I seek to correct this problem, and reveal the entire story – a story that is still being written, but nonetheless has a very dark and disturbing side to it.
In the early days, I was a big proponent of the use of “medium-chain fats” to help reverse insulin resistance and metabolic disorders which were resistant to medication or lifestyle adjustment. With time, however, I began to see a pattern that was very hard to ignore. This pattern involved people who were looking to improve energy levels, performance, cognitive ability, and much more by switching their metabolism from preferential sugar burning (glycolysis) to fat burning (beta-oxidation) – and many of them were encountering complex health issues which were difficult if not impossible for their doctors to diagnose.
MCT oil, as I will describe below, is very well suited for rapid weight loss and slight enhancements to performance and is known to deliver on such fantastic promises in many cases with short-term use. Unfortunately, quite severe complications can and do arise over time, even with intermittent use, and are almost never associated with the smoking gun that is medium-chain triglycerides.
In this article, we are going to analyze medium-chain fats, specifically capric (C10) and caprylic (C8) acids, from all possible angles and connect dots that have never been connected before. As a result, many of the concepts here may initially be met with resistance among the community of consumers at large. My only demand is that you take the time to contemplate and research on your own, using the information and references that will be shared here. Eventually, with patience and an open mind, you will come to appreciate that these risks are real and present.
As it turns out, MCT oil has a very dark side that is far worse than anyone has yet fathomed. As you will see, this is not merely my overreaction to GI disturbances people frequently experience when consuming this oil, but something much more sinister that not only changes the immune system over time, but also directly induces changes in the brain that cause neurodegeneration and the pathology for dementia.
As it stands, MCT oil is poised to become one of the primary food-based causes of neurodegeneration, and if its use is not curbed, there could eventually be a worldwide explosion in incidents of dementia. Are you surprised or even angry to hear this? Then read on.
A Brief History of MCT Oil
 The idea of using large amounts of dietary fat to affect health outcomes dates back as far as the early 1920s when the classical ketogenic diet was introduced in the form of long chain triglycerides (LCT’s). It was found back then that diets high in fat and low in carbohydrate were capable of containing the symptoms of epilepsy. For decades, this approach was used in children with epileptic / convulsive disorders, but there was a large rate of noncompliance due to the unpalatable nature of enormous amounts of long-chain fats, such as those found in nuts, cream, and butter. It was for this reason that the pediatrician, Dr. Peter Huttenlocher, and his colleagues replaced the LCT’s with MCT oil in 1971. Not only was the oil more palatable, but it also permitted a larger amount of carbohydrate and protein in the diet, making for a far more flexible lifestyle.2
The idea of using large amounts of dietary fat to affect health outcomes dates back as far as the early 1920s when the classical ketogenic diet was introduced in the form of long chain triglycerides (LCT’s). It was found back then that diets high in fat and low in carbohydrate were capable of containing the symptoms of epilepsy. For decades, this approach was used in children with epileptic / convulsive disorders, but there was a large rate of noncompliance due to the unpalatable nature of enormous amounts of long-chain fats, such as those found in nuts, cream, and butter. It was for this reason that the pediatrician, Dr. Peter Huttenlocher, and his colleagues replaced the LCT’s with MCT oil in 1971. Not only was the oil more palatable, but it also permitted a larger amount of carbohydrate and protein in the diet, making for a far more flexible lifestyle.2
That was almost 50 years ago, and since that time, MCT oil has gone from a treatment for epilepsy to a veritable boon for weight loss and reversal of metabolic syndrome. More often than not, it is combined with high-fat, low-carb diets that, at their extreme, become what is commonly known as “ketogenic”. Though the classical ketogenic diet was almost entirely composed of fat, the MCT ketogenic offshoot includes higher protein and low-glycemic carbohydrates, typically vegetables. In other words, it’s an ultra-low carbohydrate diet that’s high in fat and low in sugar and fruit, also typically excluding beans and grains.
Numerous books have appeared praising the benefits of this diet, many of which have frequently enjoyed a place on national bestseller lists. There are few that are not already familiar with the iconic “Bulletproof Coffee” and its adherents that use it for everything from weight loss to mental acuity. Businessmen and bloggers have attracted a wide following of consumers and readers of varying degrees of scientific education. They combine selections from clinical studies in scientific journals with strategies to “bio-hack” and improve body and mind, often grossly oversimplifying complex subjects in favor of attractive “sound bites” that promote health products of one kind or another. Unfortunately, one such product is MCT oil.
This dietary approach to epilepsy treatment enjoyed nearly 2 decades of success before it was abandoned in 1938 in favor of the drug Dilantin – which was able to control seizures without radical lifestyle adjustment. It wasn’t until the early 1990s that the famous Hollywood director, Jim Abrahams (of Airplane! and Naked Gun fame) brought his son, who was suffering from severe seizures, to Johns Hopkins Hospital in Baltimore. The doctors at Johns Hopkins put Abrahams’ son on the standard ketogenic diet for epilepsy, and the rest, as they say, is history. The successful reversal of his son’s epileptic symptoms was featured on NBC’s dateline in 1994, and just two years later, Abrahams created the Charlie Foundation for Ketogenic Therapies. This foundation eventually funded a successful clinical study that proved the diet’s effectiveness in treating epilepsy, and the story was adapted for the 1997 TV movie, First Do No Harm, starring Meryl Streep. Already by the mid-2000’s, the ketogenic diet had been accepted as a mainstream therapy.3
The diet got its name from the fact that the liver generates ketones in the absence of carbohydrate / glucose. It then uses these ketones to supply its energy needs, effectively “burning fat for fuel”. This fat-burning property is entirely a secondary side effect, given the original purpose was for the treatment of epilepsy. Nonetheless, it quickly became a worldwide weight loss sensation. Over the years, the benefits of “going keto” have quickly gone from promoting weight loss to doing everything from stabilizing energy, decreasing appetite, and even enabling clearer thinking. Lately, it is being proposed as a treatment for Alzheimer’s disease, and MCT oil is positioned front and center as the “medicine of choice” for improving dementia-related cognitive deficits. As I will describe further below, applying such therapies in clinical contexts may not only be premature, but even foolhardy, leading to potentially disastrous results, long-term.
Why are MCTs so popular?
One of the reasons MCT oil is so attractive for those that have adopted a ketogenic dietary template is that it can achieve ketosis without the higher 4:1 ratio of fat to protein / carbohydrate required with long-chain fats. The classic diet used nuts, cream, and butter, and for most people, it was difficult to eat enough of these food types to achieve the proper effects. Not only are MCTs more ketogenic, but they are not as caloric and, therefore, a greater variety in food selections can be made without restriction.45
Another reason medium-chain fatty acids are so popular is that instead of being stored in fat tissue, as is the case with long-chain fats, they are instead directly absorbed into the portal vein via the small intestine. Once in the bloodstream, they are metabolized in the liver to either produce ketones or generate ATP (i.e. energy) via beta oxidation.67
In animal studies, rats fed with MCTs do not gain weight as quickly as those fed long-chain fats.8 MCTs have been further shown to encourage thermogenesis (i.e. generating heat from fat-burning), whereas LCT’s are more prone to end up in fat deposits.910
Because MCTs do not require bile salts for digestion, they are also an attractive option for individuals suffering from malnutrition or malabsorption. This is an especially important consideration for those that have compromised degradation of fat.1112
Until recently, MCTs were quite rare in the Western diet with long-chain fats of 13 to 21 carbon atoms predominating. Long-chain fats are generally are more difficult to metabolize and therefore have a higher tendency to end up in fat cells. Because the body prefers to burn glucose for fuel when it is available, a diet filled with carbohydrates that convert to glucose in combination with long-chain fats, will encourage the development of adipose tissue.
Further, long-chain fats require bile salts or chylomicron formation for digestion. MCTs, on the other hand, are absorbed directly into the liver via the portal vein, completely bypassing the thoracic duct lymphatic system used by LCT’s. Nonetheless, after only five days of consuming a diet that is composed of 40% or more MCT, up to 22% of chylomicron content may include medium-chain fatty acids (with capric / C10 being dominant), indicating that at sufficient levels, MCTs follow the same metabolic pathway as LCTs in higher amounts.13
That being said, artificially isolated MCTs are not at all natural and require a multi-step process of extraction from sources such as coconut or palm oils. As a result of this extraction process, the resulting product is predominantly a combination of C8 (caprylic acid or “octanoic” acid) and C10 (capric or “decanoic” acid). Some manufacturers further refine this oil to contain only C8 which, as we will see shortly, can have detrimental effects on brain health.
MCT oil has even made its way into the Paleo diet, which centers around animal-based protein sources and higher levels of fat intake. Ironically, all oils, regardless of source, were not part of a typical Paleo lifestyle, given the technology to make them did not exist in the Stone Age. This brings into question, in general, the biological compatibility of high oil consumption for humans, from an evolutionary standpoint, especially with regard to factory-processed varieties such as MCT.
Lack of natural sources
In nature, medium-chain triglycerides are not found in great quantity, with only a few exceptions. Coconut oil is the most common source, at around 60%, but MCT content in Bacuaçu palm oil has been shown to be as high as almost 80%. Such oil is not a frequent commodity, even in developed countries, but its cousin, palm kernel oil is well known, with an MCT content of around 50%. All other sources, such as goat and sheep milk, cow’s milk, butter, ghee, etc. all have only 10-20% MCT content.
Even human breast milk contains anywhere from 10-17% MCTs, depending on pregnancy term, with preterms being on the higher end. Once we go outside the realm of dairy and coconut / palm oils, incidence of MCTs in nature are quite scarce, indeed, with beef and chicken containing not much more than 0.15%. It is also important to note that these natural sources of MCT do not contain large amounts of C8 and C10 fatty acids. Coconut oil, for example, is 45-52% lauric acid, with only 10% caprylic / C8 and 8% capric / C10. Therefore, consumption of only C8 or C10 in large amounts would not be possible without human industrial intervention.
Overhyped Benefits
 Many of the conclusions that have been drawn about MCTs and the benefits they confer are not based on a large volume of science. For example, many that promote MCT consumption for the enhancement of cognition claim it can even grow new brain cells (i.e. promote neurogenesis), but today, few if any studies would support this statement. One such study injected coconut oil into the midbrain ventricle or eyes of tadpoles, but that study was confounded by the presence of triiodothyronine (T3), which could be thought to be one of the main causes for the observed effects.14
Many of the conclusions that have been drawn about MCTs and the benefits they confer are not based on a large volume of science. For example, many that promote MCT consumption for the enhancement of cognition claim it can even grow new brain cells (i.e. promote neurogenesis), but today, few if any studies would support this statement. One such study injected coconut oil into the midbrain ventricle or eyes of tadpoles, but that study was confounded by the presence of triiodothyronine (T3), which could be thought to be one of the main causes for the observed effects.14
MCTs may help with weight loss
Regarding assertions that MCT promotes weight loss, claims in this area are also rather lacking in evidence. One study measured metabolic markers such as cholesterol, insulin, glucose, blood pressure, etc. in 49 participants and concluded after 16 weeks that MCT effects were more or less the same as olive oil.15
In another Oxford study in 2017, participants were fed 205 calories per day of either MCT, vegetable, or coconut oil. The only finding there was that MCTs were more satiating than coconut oil, but no evidence was offered that it would directly reduce weight.16
Nonetheless, there is some evidence of a difference in body composition after a month or so of MCT consumption in both obese and healthy subjects, but such results could be obtained with other methods that do not pose the same risks that will be outlined below.1718
MCTs possibly improve performance
One of the other benefits that is cited for MCT consumption is the improvement in fitness performance, but once again, the evidence here is also less than desirable. The San Diego State University study that involved endurance runners showed absolutely no statistically significant improvements in VO2 max.19
At OHSU, more favorable outcomes were observed that included notable decreases in respiratory exchange ratio, steady-state heart rate, and generation of glycolytic intermediates in parallel with elevated ketones.20 That being said, this should not embolden the public at large to embrace MCT consumption for performance gains.
Benefits Do Not Outweigh Risks
 The issue with all of the benefits associated with MCT oil is that in order to achieve such results, a significant amount must be consumed. Though at least one study proved that consumption of up to 42g of MCT per day has not shown any significant side effects, this should not be interpreted as “carte blanche” for liberal and unrestrained use.2122 Nonetheless, we see MCTs cropping up in an almost endless array of foodstuffs ranging from protein powders to chocolate bars and, of course, coffee. Not a day goes by that I don’t hear about at least one other person that has embraced the morning “bulletproof coffee” routine for cognitive sharpness, fat-burning, and weight loss.
The issue with all of the benefits associated with MCT oil is that in order to achieve such results, a significant amount must be consumed. Though at least one study proved that consumption of up to 42g of MCT per day has not shown any significant side effects, this should not be interpreted as “carte blanche” for liberal and unrestrained use.2122 Nonetheless, we see MCTs cropping up in an almost endless array of foodstuffs ranging from protein powders to chocolate bars and, of course, coffee. Not a day goes by that I don’t hear about at least one other person that has embraced the morning “bulletproof coffee” routine for cognitive sharpness, fat-burning, and weight loss.
Caprylic acid excess can induce coma
Claims have even been made that C8 oil is “superior for the brain”, given its higher ketogenic capability. At high levels, however, it has been shown to induce coma, in both human and animal studies.23 It has long been known that another C8 medium-chain fatty acid by the name of valproic acid, used commonly in children with convulsive disorders, can induce a coma if overdosed, especially when combined with phenobarbital. One study aimed to produce the same effect with C8 / octanoic acid and discovered that mole for mole, C8 was far more toxic than valproic acid in this respect.24
Valproic acid, when administered along with phenobarbital, significantly elevates brain ammonia levels. Ammonia is highly neurotoxic, and octanoic acid was shown to increase its toxicity by 12%. As a matter of fact, caprylic acid in the presence of ammonia is capable of inducing a deep coma with lower ammonia levels than valproic acid.
Comas are described in multiple stages with the last stages (IV and V) being characterized as being fully unconscious, with Stage V demonstrating complete unresponsiveness. Valproic acid can achieve these stages in 50% tested cases at 1.06 mmol in only 5-10 minutes. Octanoic acid, on the other hand, can do the same at only 0.6 mmol, a quantity 43% less than that of valproic acid.
Caprylic acid reacts with microbes in the gut
According to the NOAA hazardous materials database, octanoic acid has been found to react strongly with multiple compounds, including those which are frequently found in the GI tract of individuals with dysbiosis.25 For example, octanoic acid can react with mercaptans, such as those generated from the digestion of foods containing raffinose, to generate gases like methanethiol (methyl mercaptan).26 Together, caprylic acid and methanethiol further increase the toxicity of ammonia in the brain.27
Octanoic acid further reacts with sulfites, commonly used as a food preservative or enhancer, to generate another potentially neurotoxic gas, hydrogen sulfide. As I mentioned in my anxiety article, some bacteria, such as Pseudomonas, Citrobacter, Aeromonas, Salmonella, and Escherichia coli produce hydrogen sulfide, and though harmless at low levels, it can quickly lead to nausea, vomiting, and difficulty breathing at no less than 10-50ppm.28
Clearly, the problem here is not so much the way in which caprylic acid reacts with compounds in the digestive tract (especially those containing sulfur) but more its ability to exacerbate the neurotoxicity of ammonia – so we must consider the various reasons ammonia could become elevated.
High protein leads to higher ammonia
In healthy people, ammonia is produced as a byproduct of protein metabolism in the gut. The more protein one eats, the higher the total ammonia load is going to be. In order to keep ammonia levels down, the body uses the “urea cycle” to convert ammonia to urea and excrete it via the kidneys in urine. This is what we see measured on routine lab tests such as “base urea nitrogen” or BUN. The higher your BUN number, the more protein, in general, you have been consuming.
Unfortunately, if there is any slowdown / metabolic block in the urea cycle, or protein intake is creating more ammonia than your body can handle, there can be ammonia accumulation. As I will describe below, this ammonia is more likely to end up in the bloodstream when there is higher gut permeability – and MCTs have the distinct ability to degrade that gut barrier, thereby allowing more ammonia through. MCTs have the same effect at the blood-brain barrier and might be expected to permit more ammonia to enter the brain as well. As I will explain further below, MCTs that escape either ketogenesis or beta-oxidation are more common than many think, especially with genetic blocks in their metabolism or consumption of amounts sufficiently large to overwhelm metabolic capacity.
Brain ammonia accelerates dementia
Where else do we see aberrations in brain ammonia levels? In dementia, of course, specifically of the Alzheimer’s type, where it is shown to play a role in the morphological changes of astrocytes and glia. This is one of the many ways in which MCTs can exacerbate neurodegeneration, and volume / frequency of intake matter greatly.29
Octanoic acid’s molecular weight is ~144 g/mol, putting the amount in grams for coma induction in rats (given the presence of ammonia) at about 80mg. Considering the rats in the previously cited study weighed around 200g each, that’s a dose of roughly 400 mg / kg. Using allometric scaling, we can calculate the human equivalent dose for coma induction in a 150lb / 68kg adult to be 4.4g, in the presence of high brain ammonia levels. This means that a tablespoon of C8 oil containing roughly 14g of octanoic acid, or 14,000mg, is more than enough to pose a problem. Though the amount of caprylic acid crossing the blood brain barrier would not be as high as the amount ingested, due to dynamic variations in liver metabolism, ketone generation, and fatty acid dispersion throughout the body, in general, we can assume with fair certainty that 1 tablespoon of caprylic acid would be more than enough to reach the 4.4g threshold required for encephalopathy in the presence of ammonia (in a 150lb human being).
Again, ammonia toxicity is not going to be an issue if both your protein intake and urea cycle are optimal, and this can be said for the vast majority of people out there consuming larger volumes of MCT oil . Nonetheless, I frequently see sports enthusiasts and bodybuilders taking 6 to 8 tablespoons per day, sometimes more. While this may be utilized primarily for beta-oxidation in mitochondria-rich muscles under consistent stress, those that consume this amount that are not invoking that metabolic pathway sufficiently or that have too much protein in their diets are, in my opinion, in the line of a very clear risk. The fact of the matter is that I have seen this problem in MCT oil consumers more frequently than I would wish.
Therapeutic Uses of MCT
 One would hope that these considerations regarding ammonia, MCTs, and sulfur containing foods would be enough evidence to support abstinence from overconsumption. Nonetheless, medium-chain triglycerides continue to enjoy frequent application in the domain of neurological disorders. Contrary to popular belief, the effects are not so much related to ketones but, rather to inhibition of the glutamate-binding AMPA receptors and increase in mitochondrial biogenesis (i.e. creation of new mitochondria).
One would hope that these considerations regarding ammonia, MCTs, and sulfur containing foods would be enough evidence to support abstinence from overconsumption. Nonetheless, medium-chain triglycerides continue to enjoy frequent application in the domain of neurological disorders. Contrary to popular belief, the effects are not so much related to ketones but, rather to inhibition of the glutamate-binding AMPA receptors and increase in mitochondrial biogenesis (i.e. creation of new mitochondria).
Currently, the MCT ketogenic diet is being used worldwide to treat drug-resistant epilepsy in both children and adults.303132 Unfortunately, ambitious scientists are also proposing its use now for the treatment of diet-sensitive disorders such as Alzheimer’s disease, cancer, and diabetes.
Ketones do not mitigate seizure
In order to determine if there are valid reasons for using MCTs in these diseases, we have to examine more closely how it exerts its effects in other models such as epilepsy. Today, studies have not shown a strong correlation between blood plasma ketone levels and seizure control. In reality, ketones do not acutely block seizure at all, at least not in animal models. One study in particular showed seizure control in the complete absence of ketosis.33 This should raise some important questions.
If we look critically at the scientific literature, we see that MCTs are capable of having a direct action on seizure activity in the absence of β-hydroxybutyrate, acetoacetate, and acetone (known collectively as ketone bodies). Though the brain loves glucose and thrives on it, it can also use ketones generated by the liver to satisfy its energetic requirements.
All ketones and fats that escape metabolism for whatever reason are distributed systemically throughout the body via the circulatory system, eventually reaching the brain. Medium-chain fatty acids have shown the capability to directly cross the blood-brain barrier, reaching levels 50% of that found in blood plasma, thereby providing neurons and astrocytes directly with an alternative energy source. As such, MCTs have a wide variety of effects on brain cell energy metabolism.
For example, C8 / octanoic acid is preferentially utilized by astrocytes via beta-oxidation whereas C10 / decanoic acid stimulates the production of lactate via glycolysis, which brain cells can use as an alternative to glucose or ketones.34 It’s very interesting to note that in the presence of both octanoic and decanoic acids, brain cells prefer octanoic. This might suggest differential regulation of MCT concentrations by neurons in the brain.35
Supporters of the ketogenic diet as a therapy for epilepsy tend to adhere to the fact that ketone bodies affect amino acid metabolism, leading to alterations in the concentrations of GABA and glutamate. While GABA is inhibitory, glutamate is excitatory, so their relative concentrations are important in neuro-excitatory conditions such as epilepsy.36
Unfortunately, the evidence for ketone modulation of neurotransmitters does not stack up. β-hydroxybutyrate and acetoacetate have absolutely no effect at all on ionotropic GABA(A) receptors or glutamatergic (AMPA and NMDA) receptors and acetone and β-hydroxybutyrate have been shown to affect GABA(A) and glycine receptors only at concentrations higher than 100 mmol/L. Clearly, ketones are not the primary driver for anti-epileptic effects.37
Nonetheless, ketones have been proven to have indirect effects on neuronal excitability, and their anti-seizure mechanism has been demonstrated in animal models.383940 We could posit many explanations for this, including reversal of mitochondrial dysfunction and increase of brain adenosine levels via adenosine A1 receptors, but when we look closer at MCTs themselves, we see other mechanisms that demand further examination.4142
Capric acid is responsible for anti-seizure effects
One experiment demonstrated direct control of seizures by decanoic acid alone. It was able to single-handedly block seizure onset within only 30 minutes of application. In that same experiment, ketones were not capable of producing this effect.43
Also of note is the ability of decanoic acid to raise the seizure threshold in models of electroshock. As it turns out, it performs this feat by directly inhibiting glutamatergic AMPA receptors. This is a very important nuance for our consideration here, because AMPA receptors play a key role in the seizure pathology, and decanoic (i.e. capric acid) is capable of blocking seizure at only micromolar concentrations! Does this sound familiar? It should be clear that extremely small levels of MCTs have powerful effects on multiple systems.44
Capric acid easily accumulates in the brain
For those patients with epilepsy that observe the MCT ketogenic diet, their blood plasma levels of decanoic acid hover around 157 micromoles.45 At any level, decanoic acid crosses the blood-brain barrier quickly and without resistance, shortly after ingestion by mouth. In this way, it is capable of achieving adequate concentrations in the brain to exert the necessary effects on receptor systems.
Decanoic acid is, further, non-competitive with glutamate, enhancing its ability to inhibit AMPA receptors during synaptic over-activation (as seen in epilepsy).46 These are not new ideas, really. The antiepileptic drug Perampanel works in exactly the same manner, by inhibiting AMPA receptors, albeit at a different binding site.4748
Now for a twist in this story: the traditional MCT ketogenic diet used in epilepsy treatment contains both decanoic and octanoic acid. Of special note here is that octanoic is dominant in blood plasma on such diets, reaching concentrations of 300 micromoles or higher.49
As we look closer, we see significant differences between how C8 and C10 work in the body and brain. For example, though C8 has the same anti-epileptic actions as C10 by raising seizure threshold,50 it doesn’t seem to work on AMPA receptors at all.51 Further, unlike decanoic acid, octanoic acid does not seem to increase brain mitochondrial function via PPAR-gamma mediated biogenesis.52
Therefore, it seems highly likely that the anti-seizure control seen in the MCT ketogenic diet may be almost completely attributed to decanoic acid rather than octanoic.
MCTs for Alzheimer’s: stage matters
The question now that should be on your mind is: just how does this evidence relate to dementia? The answer is not straightforward. On the one hand, Alzheimer’s is hallmarked by a progressive inability of neurons to utilize glucose due to amyloid-beta induced insulin resistance. The ketone β-hydroxybutyrate, produced via MCT metabolism, is capable of protecting hippocampal neurons from amyloid-beta toxicity.53
Lest we jump to the conclusion that MCTs are automatically beneficial for Alzheimer’s sufferers, we need only look more deeply into the studies performed using them directly. It would appear that patients without the APOE ε4 genotype are among the very few to benefit from this treatment, showing some improvements in short-term cognitive performance.54 Such improvements, however, were generally only seen in early stages of the disease. Further, studies on mice with dementia show only improved motor function without any cognitive enhancement at all. What, then is the verdict?55
It was long ago established that glutamate AMPA receptor overactivation is one of the major contributors to the neurotoxicity of amyloid-beta.565758 Therefore, inhibition of AMPA receptors with decanoic acid has been proposed as a potential model for treating neurodegeneration in Alzheimer’s models of dementia.
Unfortunately, C10 is not preferential for its effects on AMPA-receptors and one AMPA subunit type, in particular (GluA2) actually confers impermeability to calcium, a key cofactor in glutamate-mediated neuronal excitation.5960 In fact, loss of GluA2 function effectively increases total postsynaptic calcium influx, further exacerbating inflammation and neurotoxicity. This effect, alone, is sufficient to reduce long-term potentiation and memory formation, leading to the memory loss frequently seen in Alzheimer’s patients.61
Unfortunately, loss of GluA2 is one of the first events that directly precedes the development of neurofibrillary tangles. Therefore, using MCTs for therapeutic outcomes in Alzheimer’s would not only exacerbate this effect by blocking GluA2 subunits (which are already greatly pruned down from amyloid-beta exposure), but would greatly accelerate neurodegeneration in later stages, when ammonia levels of the brain begin to rise.62
If MCT effects were limited to AMPA receptors and enhancement of mitochondrial function, it would be quite easy to embrace decanoic and octanoic acids as a therapeutic solution. Unfortunately, the dark side of these fatty acids are not limited to just AMPA receptors and ammonia interactions alone. There is a much bigger picture here that includes very sinister and troubling influences on the immune system as well.
Given the risks that I will continue to detail below, I personally believe we should seek other therapies for mitigation and / or reversal of dementia. For example, since the early 1980s, it has been known that pharmacological inhibition of ionotropic glutatmate receptors can effectively prevent seizures. The focus at that time, however, was on only glutamate receptor subtype NMDA. Therefore, NMDA receptor antagonists were the primary strategy for dealing with epilepsy. Since then, selective AMPA modulators have been under consideration as a viable alternative. The playing field, however, is riddled paradoxes.
Problems with Overconsumption
 Of all co-factors in fatty-acid metabolism, carnitine perhaps plays one of the most vital roles. It was long thought to serve only as a shuttle for long-chain fatty acids into the mitochondrial matrix63, but recent studies have proven that it plays a significant role in ketogenesis and MCT metabolism as well.
Of all co-factors in fatty-acid metabolism, carnitine perhaps plays one of the most vital roles. It was long thought to serve only as a shuttle for long-chain fatty acids into the mitochondrial matrix63, but recent studies have proven that it plays a significant role in ketogenesis and MCT metabolism as well.
You may already be quite familiar with the rhetoric, cited on keto / Paleo blogs globally, that MCTs are superior to ordinary fats given their ability to enter mitochondria without needing the “carnitine shuttle“. Long-chain fats cannot cross mitochondrial membranes without help from this shuttle, whereas MCTs may diffuse through the membrane unaided. For this reason, MCTs are often given as treatment to individuals with carnitine-acylcarnitine translocase deficiency.
The fact of the matter is that such theories are only half-true. In fact, MCTs absolutely depend on carnitine, given its ability to remove acyl and acetyl groups from inside of mitochondria which have accumulated as a result of MCT metabolism. In this way, carnitine restores intramitochondrial CoA levels and prevents disruptions to energy production (i.e. the electron transport chain).
Carnitine is slowly and inefficiently synthesized endogenously from trimethyllysine in the liver or kidney, so any degree of protein deficiency, combined with genetic aberrations in lysine metabolism, will have effects on systemic carnitine levels.64 For this reason, taking L-lysine is not a viable option for increasing carnitine levels, and for many people on ketogenic diets, it must be supplemented directly. Nonetheless, exogenous carnitine has its own side effects, not the least of which is increased production of inflammatory TMAO from gut microbiota — a subject we will table at least for this discussion.65
The positive side of this story is that carnitine’s turnover rate is also relatively slow, with only 7% of the total body pool being excreted in urine daily. That being said, because both dietary uptake and de novo synthesis from lysine are very slow processes, depletion in the presence of excess MCT consumption is a valid concern for anyone, regardless of their ability to metabolize medium-chain fats.66
Studies have shown that carnitine is produced mainly in the liver, so there is an extensive transport system to achieve whole-body distribution. It is prominent wherever there is higher density of mitochondria, such as skeletal muscle, heart, and brain. Further, carnitine levels in the blood are tightly regulated by the kidneys, so if there is any degree of kidney stress or disorder (e.g. urea cycle disruption), we can be sure that MCT-associated metabolic risks will elevate accordingly.
Most tissues contain carnitine at levels 10-100 times higher than in blood plasma, and every tissue has its own uptake rate. For our consideration, the brain’s slow uptake rate of .47-.08 nmol/hour per gram of brain tissue is significant. It means that in order to reach ~90% brain tissue saturation with carnitine, it will take roughly 220 hours.67 This means that though carnitine supply body-wide may be adequate, it is more likely to run into a deficit in the brain, especially with higher MCT consumption. Given the ease of which MCTs cross the blood-brain barrier and how, as I will describe below, capric acid accumulates in the brain (due to the absence of a brain-centric CPT1 isoform), this should be a red flag for just about anyone that consumes more than a tablespoon of this oil per day, especially if you have any degree of issue in either the urea cycle or in MCT metabolism, in general.
Carnitine depletion in ulcerative colitis
Medium-chain fats and their metabolism have broad implications throughout the body, but their relevance in gastrointestinal health is of the highest importance. Changes in fatty acid metabolism in the gut can lead to a wide variety of diseases such as IBS, IBD and even ulcerative colitis. In models of GI disease, one of the most prominent triggers is carnitine depletion, which is shown to deprive mitochondria in colonocytes of their ability to oxidize and utilize their main “fuel source”, butyrate. In this way, carnitine is a rate-limited factor for butyrate oxidation in colonic cells. Butyrate, as a short-chain fatty acid, does not require carnitine as a shuttle and may enter mitochondria directly68, but as I will describe below, MCT accumulation is capable of disturbing butyrate metabolism and blocking its effects in colonocytes.
Carnitine is transported into colonocyte mitochondria by organic cation transporters (OCTNs), and issues with these transporters has been directly correlated with risk for inflammatory bowel disease (IBD). When there is a decrease in the number of carnitine transporters or slowdown in their activity, carnitine uptake is impaired leading to less fatty acid oxidation and greater potential for injury to intestinal epithelial cells.697071
In the intestine, OCTN2 (encoded by the SLC22A5 gene) is the primary transporter, though it is shadowed by another “backup” transporter, ATB0+ (i.e. SLC6A14).7273
OCTN2 uses sodium ions to transport carnitine into intestinal epithelial cell mitochondria.74 Whenever there are issues with OCTN2, ATB0+ steps in as the primary transporter. ATB0+ has a high concentrative capacity and is thought to be important for scavenging carnitine from the distal instestine.75
If there happens to be issues in both OCTN2 and ATB0+, then there is potential for cellular damage due to fatty acid accumulation. This, in part, is one of the proposed triggers for ulcerative colitis.76 In experimental ulcerative colitis with IBD lesions, a 5-fold decrease in OCTN2 expression has been noted, with ATB0+ expression virtually undetectable. As I have mentioned above, MCTs are primarily absorbed in the small intestine, and OCTN2 is highly expressed in both small and large intestine.
In normal, healthy colonocytes, 60% of carnitine is present its free form. In ulcerative colitis, however, carnitine content is greatly decreased, and even with L-carnitine supplementation, the same level of carnitine accumulation found in normal colonocytes is not seen. If, on the other hand, those same diseased colonocytes are incubated in liposomal carnitine, there is a dramatic increase in uptake. This demonstrates that carnitine transport in ulcerative colitis is greatly impaired.77
Carnitine is absorbed primarily in the small intestine7879, however in the colon, due to a greater variety of bacteria, transporters are even more important in order to compete with microbial metabolism of carnitine.80 Once carnitine transporters have been compromised, oxidation of butyrate is also impaired. Therefore, though carnitine is not required for butyrate uptake, it is nonetheless important for its metabolism.
Butyrate has been shown to provide more than 70% of the energy required by colonocytes.81 The changes in butyrate metabolism are complex, but essentially result from a lack of required cytosolic acetyl-Coenzyme A, which is mediated by carnitine.
In any case, transporter downregulation is the result of inflammation, triggered by a decrease of free carnitine and the inability of mitochondria to properly metabolize butyrate. Therefore, the inflammation seen in ulcerative colitis is not so much a lack of butyrate uptake but, rather, a derangement of its utilization which leads to colonocyte damage.82 And this occurs, in part, as a result of carnitine deficiency.
Carnitine’s connection with ammonia
As I discussed above, dementia is hallmarked, especially in the later stages, by ammonia accumulation in the brain, and we discussed the ways that this might happen due to changes in the gut associated with protein metabolism and the urea cycle.
Valproic and octanoic acids, both C8 MCTs, have been shown to exacerbate ammonia toxicity, and it should not surprise you that one of the mechanisms by which this happens is via the depletion of carnitine. Though octanoic’s carnitine depleting properties have not been well studied, it has been established that the valproic acid metabolite, 4-en-valproic acid, directly impairs the urea cycle, thereby blocking proper ammonia elimination.
All C8 medium-chain fatty acids are capable of a similar metabolic fate, which involves hydroxylation of the omega-methyl group as a first-step reaction in omega oxidation. It is more likely to occur when beta-oxidation is impaired, as a result of carnitine deficiency, by genetic defect, or a combination of the two.
Carnitine supplementation is actually one of the antidotes for valproic acid overdose.83 Supplementation with only 1 g per day for 30 days is enough to decrease ammonia levels and raise the seizure threshold.
Given the similarities between valproic and octanoic acids, we can assume that they both follow the same routes for elimination which includes glucuronidation in the liver (50%), beta-oxidation in mitochondria (40%), and omega-oxidation (10%). The latter is the source of the toxic metabolite that inhibits the urea cycle, and it is more frequently invoked whenever either glucuronic acid has been depleted and / or there is any issue with beta-oxidation.84
To summarize, beta-oxidation of MCTs in mitochondria can be compromised by a lack of carnitine, an effect that is mediated by acyl and acetyl group accumulation within mitochondria, as opposed to the carnitine shuttle, as with long-chain fats. Further, when there is excess consumption of MCTs, the carnitine store will gradually dissipate, leading to increased extracellular fatty-acid accumulation and a greater dependency on glucuronidation and omega-oxidation for “elimination”. Many other factors can compromise glucuronidation, as we will discuss in more detail further in this article, making omega-oxidation (and its metabolic disruption of the urea cycle) a higher possibility, especially in the presence of high levels of C8.85
To be absolutely clear, I am saying that the more MCTs you have in your system, the more likely you are to deplete carnitine and shift metabolism away from beta-oxidation, leading to higher need for glucuronidation.86
In convulsive disorders, it is difficult to distinguish ammonia toxicity from seizures, themselves. Obviously, the same could be said about brain ammonia levels in dementia and the overall process of neurodegeneration. Few medical practitioners are going to look at your MCT intake to determine your risk for neurodegenerative disease. In the absence of clear clinical signs such as increased urinary metabolites, fewer still will be connecting these dots (if at all).
Damage to the choroid plexus
Even if carnitine regulation of acyl / acetyl group balance is in perfect order, there is still the possibility for inefficiency in the degradation of medium-chain fatty acids into acetyl-CoA for use in the Citric Acid Cycle. In its most severe form, this problem is known as medium-chain acyl-CoA dehydrogenase deficiency or MCAD. Through years of modeling the genes and related proteins that comprise MCT metabolism and observing symptoms and laboratory markers in people, I have come to the conclusion that there are varying degrees of this problem that may not necessarily manifest as classic MCAD. In less serious forms, there may be only minor accumulation of medium-chain length acyl-CoA compounds, and they are excreted at a rate that roughly parallels carnitine status.
As I have mentioned previously, caprylic acid easily crosses the blood-brain barrier and where there is any degree of medium-chain acyl-CoA dehydrogenase deficiency, there can be increased oxidative stress leading to intracranial pressure. As a matter of fact, one of my clients that suffers from this problem used to get significant pressure in his head after consuming any amount of caprylic acid. I didn’t make the connection that it could be the caprylic acid itself until encountering this research. For that particular client, administration of L-carnitine resolved the issue, and the scientific literature confirms the ability of carnitine to enhance the excretion of medium-chain acyl carnitines, the most prominent of which is octanoylcarnitine from C8.87
Further, in the choroid plexus where cerebrospinal fluid is produced in the ventricular system, octanoic acid has been shown to inhibit organic anion transport by disrupting mitochondrial ultrastructure. In the above referenced study, carnitine deficient rats weighing 100-150 g were injected intraperitoneally with 1 gram / kg of octanoic acid, which resulted in extensive disruptions to choroidal cells. Further, even though many cellular structures appeared to be intact, closer examination showed that the cytoplasm had filled with vacuoles, vesicles, and spaces. There were significantly fewer mitochondria, and those that survived were dysfunctional. By doing nothing more than injecting L-carnitine 30 minutes before the octanoic acid, most of these changes and disruptions were avoided.
Epithelial cells in the choroid plexus are densely numbered with mitochondria and have enzymes for glycolysis and the citrate cycle, both of which are essential to varying degrees for the supply of energy to physiological functions such as the active transport process. The choroid plexus / blood-cerebrospinal fluid barrier, is absolutely critical for the clearance of both endogenous and exogenous organic acid products via the transport system. As such, it serves as a defense mechanism preventing the disturbance of the local brain environment. This has been demonstrated multiple studies by the barrier’s ability to clear substances from cerebrospinal fluid at much faster rates than they enter the brain from the blood.
In the most severe cases of MCAD, octanoic acid completely disrupts mitochondria and all but nullifies cytochrome oxidase activity, leading to compromised energy production in the choroid plexus. This effectively disrupts the active anion transport system, leading to accumulation of potentially toxic levels of organic acids intracranially.
In late-onset Alzheimer’s disease, severe disturbances in the choroid plexus have been noted. Given that this plexus is known to take up high concentrations of carnitine in excess of blood levels, it seems a reasonable assumption that its depletion is connected with some degree of fatty-acid / acyl-carnitine imbalance. Regardless of the conclusions drawn, consuming octanoic acid in large amounts without adequate blood serum carnitine firmly increases risks, especially for those with pre-existing neuroinflammation. Further, considering the extremely slow diffusion of carnitine across the blood-brain barrier, I am not convinced that accumulation is impossible outside an MCAD diagnosis.
Even ketosis requires carnitine
Carnitine’s function does not stop at mitochondrial homeostasis for medium-chain triglyceride metabolites. There is ample evidence to suggest that ketogenesis in the liver depends primarily on the carnitine acyltransferase reaction. It has been shown that ketosis is accompanied by increased carnitine levels in the liver, leading to the conclusion that the ketogenic capacity of liver tissue is directly correlated with carnitine concentration.
This effect was demonstrated both with insulin antibodies and glucagon, effectively simulating the state of starvation which provokes ketosis. As the rate of ketogenesis increased, it was accompanied by a parallel stimulation in the oxidation of octanoylcarnitine. Therefore, in addition to being essential for elimination of medium-chain fatty acid metabolites from the intramitochondrial space, carnitine is also part and parcel to inducing ketosis, without which there will be accumulation of MCTs in blood serum and the resulting metabolic disturbances described above.88
MCTs Provoke Allergic Sensitivity
 Thus far, we have been focusing on MCT metabolism and the general risk for overconsumption. I’d now like to shift attention to a more commonly known side effect of MCTs: their ability to increase allergic sensitization to foods in even small amounts. It was originally presumed that MCTs prevent allergic reactions systemically by suppressing antigen absorption into the blood. This myth was subsequently dispelled when it was shown that medium-chain triglycerides of varying lengths stimulate absorption of antigens into Peyer’s patches, thereby invoking allergic response.
Thus far, we have been focusing on MCT metabolism and the general risk for overconsumption. I’d now like to shift attention to a more commonly known side effect of MCTs: their ability to increase allergic sensitization to foods in even small amounts. It was originally presumed that MCTs prevent allergic reactions systemically by suppressing antigen absorption into the blood. This myth was subsequently dispelled when it was shown that medium-chain triglycerides of varying lengths stimulate absorption of antigens into Peyer’s patches, thereby invoking allergic response.
Immune sensors in the gut
Peyer’s patches (PP’s) are the immune sensors in the lymphoid tissue of the intestine that transport antigens and bacteria. This tissue is actually one of the largest immune organs in the body and is known collectively as “gut-associated lymphoid tissue” or GALT. It discriminates between dangerous pathogens and harmless bacteria in the gut and is so large, in fact, that is said to contain as much as 70% of the body’s entire immunocyte content.89
PP’s consist of collections of lymphoid follicles surrounded by a follicle-associated epithelium (FAE). The FAE mediates exchanges between the GALT and the internal luminal environment. Specialized cells called microfold- or “M”-cells transport antigens and bacteria from the lumen toward immune cells that are responsible for inducing or limiting immune response.
PP density reaches its peak between the ages of 15 and 25 and subsequently declines as life progresses, making individuals in that age group at increased risk for MCT-induced antigen sensitivity.90 Nonetheless, other age groups are at risk as well. For example, Peyer’s patch numbers in the ileum are at their highest from the age of 30-40.91 In the small intestine there is an average of only 60 PP’s at week 30 of gestation and this number gradually increases to 240 by the time a person reaches puberty!
What is most important for our consideration here is how highly “plastic” PP containing FAEs are and how they may be modulated in different ways, depending on the microbial content of the gut. As a matter of fact, M-cell numbers have been shown to increase in pathogen-free mice as soon as they are migrated to “normal” housing conditions.92 As such, even transient exposures to pathogenic bacteria (e.g. Streptococcus or Salmonella) is capable of increasing M-cell numbers within the FAE.9394 In this way, the FAE can rapidly adapt to changing conditions in the gut lumen, depending on host immune status and stimulation from bacteria.
The critical point I would like to make here is that paracellular permeability is regulated in the FAE quite differently than in intestinal mucosa. Increased expression of tight junction proteins such as claudin-3 and occludin can be found in the FAE, lending to its ability to prevent tight junction opening associated with gut permeability. As I will explain, MCTs are capable of altering these proteins and directly changing tight junction structure, thereby increasing translocation of antigens, bacteria, and viruses.9596
Altered immune tolerance
Immune tolerance of commensal (i.e. non-pathogenic bacteria) is a unique property of the gastrointestinal mucosa. It should be obvious that anything that would enhance sensitivity to such bacteria could prove ultimately devastating to gut health.97 Tolerance to commensal bacteria is encouraged in the gut mucosa via antigen-specific T lymphocytes that suppress immune response, also known as T-regulatory cells or “Tregs”.
Tregs induce “antigen-specific suppression” of both cellular and humoral immune responses. Without mucosal tolerance, there is increased risk for inflammatory damage to the gut mucosa as a result of “hyper-reactivity”. This delicate balance only works when the immune system recognizes commensal bacteria from previous exposures. For example, it has been demonstrated that elimination of protein from the diets of mice leads to an underdeveloped GALT and low levels of secretory IgA. This is an environment ripe for “hair-trigger” allergic response. Suddenly re-introducing protein to the diets of such mice, indeed, produces antigenic responses.98
In fact, this is exactly how food hypersensitivity such as celiac disease develops. In celiac disease, there is a lack of Treg-mediated suppression of immune response to wheat-derived proteins such as gluten. As a result, eating gluten can produce a hyper-reactive response leading to severe inflammation and damage to the intestinal epithelia. Now, imagine if you will how immune sensitizers such as MCT can produce such a reaction in individuals without celiac disease that are eating pro-inflammatory proteins such as gluten or gliadin.99100
Increased risk for pathogen translocation
It should be abundantly clear that MCTs increase antigen uptake into Peyer’s patches, providing them access directly to the internal immune cascade. In this way, PPs also open the door for bacteria, viruses, protozoa, and even prion. Escherichia coli, Yersinia, Mycobacterium avium paratuberculosis, Listeria monocytogenes, Salmonella typhimurium and Shigella flexneri have all been reported to invade their hosts by adhering to FAE M-cells. What then, can we say about such pathogens in the presence of MCT? Should we feel confident about the safety of MCT consumption on an empty stomach (free of potential food antigens) if such invaders are lurking in waiting down in the gut? Would even the MCT content of pure coconut oil be enough to provoke a reaction? The answer should be obvious. Yes, even coconut oil can induce this reaction in those with a hyperactive immune system.
In all situations involving pre-existing gut infections, regardless of magnitude, translocation rates for these pathogens is increased.101 The hard truth here is that I have noted such infections in multiple clients, all of which had been consuming MCT or even coconut oil to one or another degree. Many of them have developed systemic inflammation from endotoxin translocation, and I would go as far to say that MCT-induced inflammatory response to those pathogens was responsible for the gut permeability that allowed that translocation. It is of utmost importance that we understand the interactions of such pathogens with Peyer’s patches and how MCTs amplify antigenic response. It should be more than abundantly clear what the implications could be for the brain, which is protected by both the blood-brain barrier and the choroid plexus.
H. Pylori and viral virulence
GI effects of MCT are not limited to the intestinal tract. In the stomach, H. Pylori has been shown to induce gastritis via Peyer’s patch mediated translocation. In fact, mice lacking Peyer’s patches do not contract gastritis from H. Pylori. We have already established that MCTs induce their effects locally on contact, so it is not at all a far-reaching conclusion to assume it could increase H. Pylori virulence directly in the stomach as well, on it’s way down to the small intestine.102
Let’s not forget the implications for viruses and their effects on the human nervous system. It may surprise you to discover that M-cells also mediate transport of Poliovirus and HIV type 1.103104 Fortunately, poliomyelitis has not been a significant public health concern for quite some time, but it should nonetheless be of interest that it infects humans primarily via the oral route, and PPs are the primary sites for viral replication in the gut.105 HIV-1, on the other hand, gains access to CD4+ T-cells by adhering to M-cells and translocating across the mucosal barrier of the intestinal or genital tracts.106 In this way, MCT consumption also increases risk of orally contracted HIV-1 infection, and its further use in HIV-compromised individuals would only accelerate its invasive potential.
Confusion between friend and foe
Another important question we should be asking with regards to MCT immune effects is how commensal bacteria are distinguished from pathogenic microbes. The answer lies in patterns present on the surfaces of both commensal and pathogenic bacteria called PAMPs or “pathogen associated molecular patterns“. Host cells contain pathogen recognition receptors that bind to PAMPs, thereby relaying signals which distinguish whether the microbe is “friend or foe”.
The most common PAMP receptors are known as Toll-like receptors (TLRs) and the Nucleotide oligomerisation domain (NODs), which are highly expressed in epithelial and dendritic cells. While TLRs are primarily sensors outside the cells, NODs act within the cell. In other words, we have guardians on watch both inside and outside of our cells. NOD2, in particular, is adept at recognizing muramyl dipeptide present in the bacterial cell wall of both gram-positive and gram-negative bacteria. It should come as no surprise that genetic variants in NOD2 often lead to GI diseases such as Crohn’s.107108
Germ-free animals with underdeveloped GALTs have been shown to be resistant to Chron’s disease. This would imply, once again, that bacterial sensors in Peyer’s patches could play a role in over-reactive host immune responses.109 Indeed, mice missing the NOD2 gene are noted for their GALT hypertrophy and hyperplasia (i.e. overdevelopment). In other words, they are more resistant to hyper-immune responses in gut lymphoid tissue.110111 More than several studies have indicated the potential role NOD2 deficiency plays in the GALTs response to commensal bacteria.112
When given large doses of antibiotics, GALT overdevelopment is halted in NOD2 deficient mice. In such cases, when commensal flora is re-introduced, it produces strong immune stimulation in NOD2 deficient Peyer’s patches. This leads to elevations in CD4+ T cells, higher levels of inflammation, and a high rate of permeability for antigens and bacteria.113 In other words, NOD2 deficiency results in higher reactivity to commensal bacteria and a decreased ability to suppress colonization of pathogenic bacteria.114 In this way, NOD2 plays a key role in the regulation of the interaction between Peyer’s patches and gut flora.
When NOD2 is optimally expressed, GALT development will be balanced, and there will be less inflammatory response to bacteria. On the other hand, if there are any genetic variations in NOD2 expression, this can be expected to lead to higher levels of both CD4+ T-cells and M-cells in the FAE with increased Th1 pro-inflammatory cytokines. In the presence of MCTs, NOD2 deficiency would exacerbate Peyer’s patch-mediated immune stimulation and further increase pro-inflammatory signaling. This would further manifest as higher translocation of pathogenic bacteria via PPs.115116 In this way, MCTs are capable of gradually sensitizing a host to hyper-reactive immune responses to its own beneficial bacteria. This has profound implications for those that combine MCT-laden diets with probiotic supplementation (or foods high in commensal bacteria). Naturally, the risk increases depending on NOD2 genetic status.
Without optimal NOD2 expression, there will be lower immune tolerance and elevated risk for both paracellular and transcellular permeability. This translates to very “bad things” leaving the gut lumen and entering the bloodstream. Crohn’s disease, in particular, is marked by NOD2 mutations that lead to a deficiency of Tregs in the lamina propria of the colon.117 As such, Chron’s disease lesions are often quite close to Peyer’s patches, and it can be presumed that such lesions are the result of an inappropriate immune response to gut bacteria, regardless of whether they are friend or foe. Indeed, Crohn’s disease has been shown to be most prominent in the ileum where there is an abundance of Peyer’s patches.118
As stated above, we see a peak in Peyer’s patch numbers between the ages of 15 and 25. Is it any coincidence that Crohn’s disease is generally more prevalent in that particular age group? Ileal Crohn’s disease is all but unheard of among young children and seniors.119120121
The obvious role Peyer’s patches play as an interface between gut flora and immune response becomes all the more pertinent when discussing MCT modulation allergic sensitivity. It may seem irrelevant to our discussion of dementia, but do not forget that multiple studies already exist demonstrating the correlation between altered gut microbiota, amyloid formation, and the pathogenesis of Alzheimer’s disease.122 Consider how modulation of immunity in the GALT could influence this dynamic over time and the ways described above in which MCTs could profoundly exacerbate or even trigger that process. If you are still not convinced, then please read on.
Allergic inflammation and anaphylaxis
Highly allergenic foods such as peanuts are known to contain high levels of triglycerides, but it was never presumed that they could play a role in allergic reactions directly. It was later found that triglycerides increase antigen absorption, thereby elevating sensitivity to them and, in some cases, even causing anaphylaxis, the latter of which happens via both IgG and IgE mediated mechanisms.
MCTs play a unique role in T-helper 2-type (Th2) allergic responses via their stimulation of thymic stromal lymphopoietin (TSLP). TSLP is a cytokine produced by the intestinal epithelium which effectively promotes the induction of allergic responses in various ways.
When it comes to allergic response in the gut, the intestinal epithelium plays the most significant role. Antigenic material does not have access to the lamina propria and beyond unless the intestinal epithelial cells allow them through. Low levels of gut permeability can, in fact, increase tolerance to antigens via gradual exposure – similar to the way allergy shots work. On the other hand, the more permeable the gut barrier becomes, the more antigen it lets through and, thereby, the more severe allergic sensitivity becomes.123124125 It is interesting to note that when combined with long-chain fats, the chylomicrons used for LCT absorption into mesenteric lymph prevent basophil activation and mitigate the allergy-sensitizing response MCT would otherwise provoke.
As I have already described above, MCTs are not only capable of exacerbating allergic reaction and antigen absorption into Peyer’s Patches, but it also has the capability of inducing transient mucosal damage and gut leakiness which can lead to further translocation of antigens from foods (and even bacteria and parasites) into the bloodstream.126 In the above-mentioned study, when LCT’s in peanut butter were replaced with MCTs, there was a significantly elevated Th2-mediated allergic response followed by absorption of the peanut antigen into Peyer’s patches. Anecdotally (i.e. if you follow forums and blogs where MCT-use is popular), you will discover that this same reaction has been reproduced in many different contexts that include (but are not limited to) eggs, nuts, various vegetables, and even meats, to varying degrees. Few suspect that MCT could have been at the root of an acquired food allergy.
With chronic MCT consumption, we see extremely elevated expression of TSLP mRNA in the both the jejunum and intestinal epithelia. This increase in allergic sensitivity happens almost immediately, but can increase to clinically significant levels in as little as 3 weeks of constant exposure. This TSLP upregulation is not observed in areas of the GI tract that are not exposed to MCT, indicating this is the result of direct contact of MCTs with the gut lumen.
After the sensitization with MCT has occurred, there is a subsequent uptick in mast cell degranulation releasing progressively more histamine both locally in the gut and into the bloodstream. This mast cell degranulation in response to MCT may be effectively blocked by preventing receptor-mediated IgG responses.127 Considering these facts, it’s very clear that ingestion of in MCTs to any degree, with or without a food substance to which an allergy already exists, there is the potential for increased gut barrier permeability, absorption of antigen into Peyer’s patches, increased antibody production, and in extreme cases, the potential for anaphylaxis from any antigen to which one has been sensitized by MCT.
As I have already pointed out, this sensitization is not limited to foods but also includes both commensal and pathogenic bacteria – literally anything that may be found in the gut. And if you have been reading carefully, you will remember that MCT also reacts with the gases produced by gut microbes. These gases also have the potential to directly increase bacterial pathogen absorption and sensitization leading to greater gut permeability and absorption of gut endotoxin into the bloodstream. This is the beginning of a cascade that cannot only trigger autoimmune disorders, but also progress to neurodegeneration, considering that these same processes have also been observed directly at the blood-brain barrier. This, alone, should dissuade anyone from consuming MCT at all but unfortunately, antigen sensitization is only beginning of the problem.
While it could be said that mixing MCTs with long-chain fats could potentially mitigate this cascade to some degree by inhibiting antigen access to mast cells, basophils, and dendritic cells that are responsible for immune stimulation, there are other mechanisms involved here outside of the immune effects I have mentioned that are equally as detrimental to health.
The majority of mast cells in the GI tract reside in the lamina propria of the upper gastrointestinal tract, so allergic sensitization can happen very early on in the digestive process. Knowing that MCTs are primarily absorbed in the small intestine, this should give you pause. Further, if there is any degree of deficiency in chylomicron production, secretion, transport, and clearance, which can be a genetic issue, there will already be an increased risk for food allergy with chronic administration of MCTs, with or without long-chain fats.
TSLP – master allergy inducer
TSLP is a master cytokine responsible for the induction of allergic responses.128129 It is produced primarily by epithelial cells and plays a critical role in the regulation of immune responses via the maturation and activation of dendritic cells, lymphocytes, basophil precursors, and fibrocytes.130131132133
It is common to see increased TSLP production at the sites of inflammation such as those seen in severe asthma, allergic rhinitis, and even atopic dermatitis.134135 In mice that are deficient in TSLP receptors, there is close to no incidence of asthma or allergic dermatitis. We can easily extend this to include the intestinal epithelium, which has already been demonstrated to express TSLP mRNA in the presence of antigen.136137
To put it simply, wherever there are epithelial cells, there is the potential for TSLP-induced allergic reaction. TSLP mRNA levels were shown to increase significantly, for example, by doing nothing more than painting caprylic acid on the ear lobes of mice.
Octanoic acid’s effects on dendritic cells may also be seen in the skin, where it acts as an immune sensitizer. Dendritic cells resident in skin migrate to skin draining lymph nodes where they present sensitizing proteins to naïve T cells, which are dispersed into peripheral circulation. Repeated exposures to skin sensitizers such as octanoic acid leads to chronic allergic contact dermatitis, in part induced by upregulation of pro-inflammatory genes such as ATF3 and chemokine CXCL8.138
MCTs Accelerate Neurodegeneration
 One of the issues with MCT oil containing both decanoic and octanoic acids is that CPT1 (carnitine palmitoyltransferase I), required for beta-oxidation of decanoic acid / C10 in mitochondria, is poorly expressed in the brain. This leads to the potential for decanoic acid to accumulate in the brain, and this effect is enhanced in the presence of C8.
One of the issues with MCT oil containing both decanoic and octanoic acids is that CPT1 (carnitine palmitoyltransferase I), required for beta-oxidation of decanoic acid / C10 in mitochondria, is poorly expressed in the brain. This leads to the potential for decanoic acid to accumulate in the brain, and this effect is enhanced in the presence of C8.
Both C8 and C10 have been shown to accumulate in the plasma of individuals consuming large amounts of these fatty acids, such as those on the MCT ketogenic diet.139140 As we’ve already discussed, C10 has proven its ability to inhibit AMPA receptors, thereby providing high potential for seizure control. Therefore, having higher serum levels of C10, with or without C8, may be desirable for those under treatment for epilepsy.141 Nonetheless, mouse studies have shown significant accumulation of C10 in the brain,142143 and this accumulation is variable, depending on the C10 / C8 ratio of the oil used.144
One of the questions that had researchers puzzled initially was how C10 could accumulate at all, since it was presumed that MCTs are rapidly oxidized and converted to ketones (mostly in the liver). If beta-oxidation and ketone production were efficient, how then could sufficient levels of C10 be achieved in the brain to produce anti-seizure effects?
Multiple studies have confirmed that C10 quantity must reach at least 250 micromoles to promote mitochondrial biogenesis, improved antioxidant status, and inhibit AMPA receptors.145146147 This appears to be achievable through ordinary oral administration.148149
We’ve already discussed how medium-chain fatty acids are catabolized by beta-oxidation, resulting in acetyl-CoA formation. At that stage, they can either be further metabolized to ketones or enter the Citrate cycle for energy production.150151 When MCTs are administered with glucose, the brain has been shown to prefer glucose as its primary neuronal energy source.152153154155 Nonetheless, even though C8 and C10 have lower oxidation rates than glucose, they are taken up by neurons even in its presence.
C10 has a significantly lower beta-oxidation rate than C8. As it turns out, C10 has been found to employ the carnitine shuttle to improve beta-oxidation potential! For reasons stated above, carnitine metabolism may be disturbed in certain individuals which can lead to further potential for C10 accumulation. Again, this effect is amplified in the presence of C8.156 Even with low to zero level concentrations of C8, C10 beta-oxidation is still quite slow. This has led to the conclusion that one of the reasons the MCT ketogenic diet is so effective at inhibiting seizures is because C8, when given together with C10, promotes C10 accumulation and, thereby, C10-specific anti-convulsant effects. In fact, such effects might decrease if carnitine were supplemented in parallel. Does this sound like a slippery slope to you?
A thorough review of CPT1 activities in the brain reveals a brain-specific isoform, known as CPT1c, but multiple studies have confirmed it does not demonstrate a relevant level of enzymatic activity.157158159160161 Even in astrocytes, C8 is clearly ketogenic, whereas C10 does not display this capability at all.162
Blocks in MCT metabolism cause brain damage
Once again, I’d like to point out that any degree of dysregulation in medium-chain fatty acid metabolism, regardless of whether or not it is full-blown MCAD, can increase the possibility of C10 accumulation in the brain. This is not to say that C8, alone, is not without issue, as should be apparent based on our discussion thus far about immune regulation. Nonetheless, it is important to understand what happens in the human brain, overloaded with C10, in contexts that do not involve epilepsy.
C10, in excess, has shown to have a wide range of neurological effects, not the least of which is the inhibition of mitochondrial cytochrome C activity by as much as 30% and complex II-III activity by 25%. These are core mitochondrial “components” for the production of ATP (energy) in neurons.163
While octanoic acid in sufficient amounts can produce coma, at lower levels (in rodent studies), the effect is decidedly narcotic, in part via inhibition of mitochondrial ATP-ase activity. This effect has been attributed to the monocarboxylic acid carrier for octanoic acid in the blood-brain barrier.164165166167 As already mentioned, both C8 and C10 can disrupt organic acid transport in the choroid plexus, leading to accumulation of a wide array of organic acids, including medium-chain triglycerides.168
Even when antioxidants such as reduced glutathione (GSH), ascorbic acid, or vitamin E are administered to MCAD subjects, they fail to prevent the inhibitory effect of decanoic acid on creatine kinase in mitochondria. As a matter of fact, ascorbic acid and vitamin E both decrease the activity of creatine kinase, further exacerbating ATP / energy deficit.
Citrate is transported across the mitochondrial membrane by the tricarboxylic carrier and generates carbon dioxide after catalytic reactions by aconitase and isocitrate (in the Citrate cycle). Decanoic acid induces markedly lower CO2 production, demonstrating its ability to directly block the Citrate cycle. Octanoic has similar inhibitory effects on carbon dioxide formation from acetate, blocking acetyl-CoA synthase directly in the brain.169
This effect is more pronounced with glucose than with acetate / ketones. Inhibition of glycolysis (i.e. production of energy from glucose) in the brain, another biomarker of Alzheimer’s disease, has been demonstrated with both octanoic and decanoic acids, and multiple studies have further confirmed that MCTs are capable of inhibiting key glycolytic enzymes such as hexokinase, phosphofructokinase, and pyruvate dehydrogenase.170171172 Therefore, it is quite clear from the literature, that excess MCTs in the brain, especially decanoic acid, can block not only the Citrate cycle (i.e. CO2 production) but also mitochondrial respiration, and this effect is amplified in accordance with individual metabolism of medium-chain fatty acids.
Decanoic acid, which accumulates more rapidly in the brain than C8, is responsible for mitochondrial respiratory chain inhibition, while octanoic primarily affects glucose metabolism. To explain this in laymen’s terms, inhibition of mitochondrial respiration by excess decanoic acid parallels the decline in ATP synthesis (i.e. neuronal energy) that accompanies neurodegenerative disorders such as Alzheimer’s, ALS, and Leigh disease. Such neurodegenerative disorders also show lower mitochondrial Complex II and III activity, both of which are also inhibited by C10.173174175176
To be very clear, all it takes to induce a potentially deadly crisis in those with MCAD is 0.6 and 0.2 millimoles of octanoic and decanoic acids, respectively, in blood serum.177 Once in the blood, they cross the blood-brain barrier with ease.178 Octanoic acid has been shown to compromise brain energy metabolism both in vivo and in vitro in relatively low amounts when MCT metabolism is even slightly compromised. This creates the perfect storm for potentially severe neurological symptoms due to changes in mitochondria.179180 Exposure of brain tissue to an excess of both octanoic and decanoic acids can cause lower ATP-ase activity in synaptic plasma membranes leading to impaired mitochondrial respiration in both liver and brain.181182
It is extremely important to emphasize here that these issues arise on a very broad spectrum from no symptoms at all to full-blown metabolic shutdown. If you are familiar with my theories on SNPs and genome-wide studies, or worked with me as a researcher or client, you will know that I believe MCAD deficiency may manifest in ways that have not yet been clinically correlated to DNA mutations. In fact, I would go as far to say that it may be subclinical and virtually undetectable in many people – until, of course, a stressful event disturbs metabolic balance. Can you think of anyone you know that was perfectly healthy until “that breakup” or “that bad cold”, at which point everything began to fall apart? Those are examples of that fine line we all walk, depending on genetics, environment, and lifestyle. That being said, we need to look critically at things that can interfere with medium-chain acyl-Coenzyme A dehydrogenase (MCAD) activity, outside the realm of genetic defects. It is my belief that the risks in this area are much higher than has been acknowledged to date.
Here, I will quickly list some of the ways in which the ACADM enzyme (acyl-Coenzyme A dehydrogenase, C-4 to C-12 straight chain) may be inhibited along with relevant references:
- Choline deficiency and NAFLD183
- Lipoic acid deficiency combined with ketogenic diet184
- Persistent (POP) organic pollutant exposure185
- Excess estrogen186
- Overconsumption of alcohol187
- High lactic acid to pyruvate ratio188
- Excess ketones189
- Lipopolysaccharides (LPS) / endotoxins from gut pathogens190
- Oleic acid together with animal fat / lard (i.e. palmitic acid)191
- Hypoxia of any degree192
- Testosterone deficiency193
Increased risk for stroke
TSLP, as an allergic stimulator, is not just confined to the gut. It may be found in skin, lungs, and thymus, but most importantly – it is also present in the brain. In fact, receptors for TSLP in the brain are most abundant in neuronal cell membranes and in the cytoplasm of neurons, astrocytes, and microglia. They play a major role during ischemic stroke in the cortex via their interactions with STAT.194 Given the WHO has predicted ischemic stroke will be at epidemic levels in the 21st century, we can’t help but ask what can be done to mitigate this problem.195
Similar to dementia, stroke is triggered as a result of excessive neuroinflammation. One could even say that if you have risk for one, you are also at risk for the other.196 Microglial cells in the central nervous system are capable of differentiating into noninflammatory dendritic cells, and TSLP is produced by epithelial cells in the choroid plexus as well as the spinal cord’s astrocytes.197198
Anything that can influence TSLP levels in the brain and its nervous system can lead to excessive neuroinflammation, such as that seen in dementia and stroke. In fact, neuroinflammation is actually a protective mechanism against brain insults which is automatically activated in response to either vessel occlusion or, in the case of Alzheimer’s, amyloid-induced oxidative stress.199200
STAT-mediated up-regulation of protein expression is required in order to induce inflammatory processes.201202 STAT3 in humans and STAT5 in mice have been shown to play critical roles in this cascade. For example, lipopolysaccharide that translocates into the blood stream due to gut permeability can directly activate STAT proteins. Should endotoxin cross the blood-brain barrier (due to, for example, a compromised choroid plexus), then it will be able to invoke neuroinflammation by the same STAT-mediated cascade.203204
It is specifically this STAT-mediated mechanism that induces the “protective” neuroinflammatory process in the brain in the early stage of ischemic stroke, and it is thought to mitigate potential brain damage. During this process, multiple pro-inflammatory cascades are invoked, including the activation of TSLP, which triggers dendritic-cell mediated Th2 / allergic-type inflammatory responses. Not only does TSLP induce the maturation of DCs, but it also, via production of Th2 cells, provokes the activation of STAT.205
TSLP expression in the epithelial cells of the choroid plexus and astrocytes regulate the survival and function of Th2 cells in meningeal / perivascular network of dendritic cells.206 Therefore, it should come as no surprise that higher levels of both TSLP and its receptor exist in neurons and gliocytes to regulate neuroinflammation in the early stages of ischemic stroke, accompanied by elevated STAT protein interactions.
Knowing this sheds new light on our examination of what excess MCTs in the brain are capable of, considering their ability to directly induce TSLP. It should be no stretch of the imagination to guess what could happen in cases of a compromised blood-brain barrier or choroid plexus, both of which could be directly induced by MCT effects on transporters and tight junction proteins, the latter of which we will discuss in greater detail below.
MCTs provoke autoimmunity in the brain
All of the points I have been sharing here are exponentially more relevant in individuals that have any degree of autoimmunity. Any aberrations in dendritic cell function can lead to a loss of ability to maintain tolerance to antigens and result in “autoreactive” immune responses to self-proteins. This can only exacerbate inflammation, especially neuroinflammation. In fact, dendritic cells have been implicated in multiple neuroinflammatory disorders including multiple sclerosis, HAM/TSP, Alzheimer disease and prion-associated diseases.
Immunoglobulins, also known as autoantibodies, play critical roles in a wide array of autoimmune diseases.207208209210 Self-reactive T cells are also implicated in autoimmunity.211212213214215216
In individuals with healthy immune systems, autoantibody-expressing B cells that bind to self-proteins are automatically tagged and forced to undergo apoptosis (i.e. programmed cell death). Sometimes, these cells escape negative selection, move out of bone marrow, and into other areas of the body.
T cells also undergo a process of selection, but it is more complicated. In brief, T cells that recognize self-proteins are preferentially selected, and are then eliminated by virtue of the degree to which they react to self-proteins. Nonetheless, similar to B cells, certain auto reactive T cells manage to escape this process and reach other areas of the body where they can become activated and proliferate.
There are a number of “backup plans” in place to ensure that auto reactive T cells lurking in the background do not become activated. Such plans include anergy, clonal deletion by activation, induced cell death, and antigen sequestration.
Though the development of autoimmune diseases often revolve around certain inherited genes that make individuals more susceptible to environmental triggers, thereby promoting T cell reactivity, certain conditions can also increase the likelihood of silent, auto-reactive T cells moving into action. Such conditions include infections, especially of the viral kind. Various products derived from viruses, for example, have the ability to mimic host proteins, resulting in “immune confusion” and targeting of self-proteins using antigen-directed immune mechanisms. Dendritic cells play a major role in this process by policing their environment and waking up autoreactive T cells in response to certain inflammatory signals or infections.
Dendritic cells, also called “Langerhans cells”, were first described by Paul Langerhans at the end of the 19th century. Steinman and Cohn later coined the term “dendritic cells” in 1973, and Steinman eventually went on to win the Nobel Prize for discovering their immune effects. They were found to absorb, process, and present antigens directly to T cells as a part of both the primary and secondary immune responses.217218219 Today, four types have been characterized: interstitial, Langerhans, myeloid, and plasmacytoid.
Although dendritic cells are known to induce T regulatory cells that suppress autoimmunity,220, they have nonetheless also been seen to interact with T cells, initiating a pro-inflammatory cascade in resident tissue.221 For example, in inflammatory diseases such as psoriasis, DCs have been shown to accumulate inside lesions.222 One study even demonstrated the ability of certain dendritic cells to exert pro-inflammatory effects via the production of nitric oxide and TNF-alpha. Further research in this area has shown that DCs can infiltrate the skin of patients with psoriasis, become activated, and produce interferon-alpha.223224
Clearly, DC functions are very much dependent on their local environment and the other processes that are happening around them.225 It is therefore important to note that dendritic cells of the brain are characterized by their capability to stimulate T cells, exacerbating neuroinflammation, and eventually causing neuronal death. This very same pattern has been seen in models of autoimmune diabetes, where DCs infiltrate pancreatic islets and lead to beta-cell destruction.226
In the early days, it was presumed that dendritic cells did not have access to the central nervous system. It wasn’t until 1996 that they were first identified directly in the CNS.227 This is a critical finding, as even if antigens in the central nervous system are able to evade adaptive immunity in the brain, they may still be targeted by T cells, resulting in widespread neuroinflammation.228
As described above, these very T cell responses are induced by none other than the dendritic cells themselves.229 As a matter of fact, DCs actually accumulate in the central nervous system parenchyma during immune activity.230 It is precisely in this manner that densely packed dendritic cell clusters are sufficient to present antigen to myelin, leading to the neuroinflammation that is commonly seen in multiple sclerosis.
DC-SIGN positive cells have been seen in close proximity to invading T cells in both acute and chronic active lesions in multiple sclerosis. DCs have even been spotted directly in the blood-brain barrier and meninges.231 These SIGN+ DC-cells are also in normal brain tissue, leading to the conclusion that antigen presentation may very well happen directly at the blood-brain barrier, provoking the entry of T cells into the central nervous system.
Many of the dendritic cell mediated inflammatory processes that I’m describing here are often happening for decades before any noticeable symptoms arise. Alzheimer’s related neuroinflammation, for example, doesn’t present symptoms typically until the age of 50.232
As we’ve already discussed, beta-amyloid is the most destructive component in the pathology of Alzheimer’s disease, and neurons are vulnerable to one degree or another based on the level of complexity in dendrite structure. Many of the early studies on Alzheimer’s pathogenesis focused on microglia and their ability to secrete pro-inflammatory cytokines, leading to neuronal injury.233234235
Class A scavenger receptors are responsible for the uptake of beta-amyloid in microglia, but degradation is minimal.236237 In fact, amyloid has shown the ability to evade immune recognition by failing to activate antigen presenting cells such as DCs.238 In the presence of excess MCTs such as decanoic acid, however, we see a different cascade.
As I have described above, MCTs evoke TSLP expression, which primes and conditions DCs. In this environment, when amyloid beta generates ROS in neurons, a stress signal is issued via heat-shock proteins, “alerting” DCs regardless of whether or not antigen is present. So we now have overactive DC-mediated activation of both autoreactive T cells, and by virtue of their damage to the neurons themselves, antigen-type reactivity to self-proteins. What could be an otherwise slow process taking several decades to unfold can, under the right circumstances, accelerate exponentially. Why aren’t we hearing more about this effect in studies? Because there is close to no cross-talk between the scientists studying neuronal effects of MCTs in models of epilepsy and neurodegenerative disease and those studying the immunological aspects of those diseases. Only now are these dots being connected, perhaps for the first time.
Clearly, even in the absence of autoimmunity, the degradation of neurons induced by amyloid proteins can induce stress signals that lead to rampant neuroinflammation, and adding medium-chain triglycerides to this milieu is only going to throw oil on the fire. This problem becomes exponentially more pronounced if antigens such as endotoxin (or other unwanted molecules) are present as a result of a compromised blood-brain barrier and / or choroid plexus.
The idea that dendritic cells may be recruited into the central nervous system remains a controversial topic. If this were the case, however, then DCs from other areas of the body could make their way into the CNS and with them, pathogens and endotoxins.239 No matter how you look at this, there is a clear need to be exceedingly careful with anything that can provoke a hyper-reactive immune response, especially in the brain.
MCTs encourage memory loss
Recent clinical trials have demonstrated that frontotemporal dementia is characterized by a wide array of changes in neurotransmitter systems such as serotonin, dopamine, GABA, and glutamate. Of these, glutamate is considered to be a critical player in its development240 Hypofunction in N-methyl D-aspartate (NMDA) and α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) receptors has been shown in animal models, and in humans, we have seen a loss of glutamatergic pyramidal neurons across the brain, especially in the frontal and temporal cortices.
Frontotemporal dementia is characterized by multiple abnormalities in behavior along with impairments of language and executive function. It is one of the most common neurodegenerative conditions after Alzheimer’s disease, although both pathologies could potentially be combined into a single category due to the similar way in which they develop.241
Much of the scientific literature is focused on genetic mutations which predispose individuals to risk of dementia, including Microtubule Associated Protein Tau (MAPT), known to cause tau accumulation, and Granulin (GRN), a protein that aggregates in tissues during inflammation.242 Genetic mutations aside, early diagnosis and continued assessment of neurodegeneration associated with dementia has been a considerable challenge.243244
In 2017, anti-AMPA GluA3 antibodies were identified in blood serum and cerebrospinal fluid from patients with frontotemporal dementia. This further supports my hypothesis that autoimmune mechanisms, in part mediated by TSLP and dendritic cells, is at the root of many cases of dementia.245 Indeed, other scientists have been thinking along the same lines, and just a couple of years ago a theory was posited that dementia could be treated by either modulating the immune system or supporting optimal glutamatergic receptor density and signaling, similar in approach to the existing dopaminergic and cholinergic therapies for Parkinson’s and Alzheimer’s, respectively.246
Modulation of glutamatergic signaling is challenging, as overactivation leads to brain ischemia, epilepsy, and neurodegeneration, while underactivation results in cognitive impairment and loss of memory. As I have clearly outlined thus far, MCTs can both trigger immune responses in the brain leading to overactivation of these receptors and, as the pathology grows, inhibit signaling further (especially when there are higher levels of C10). Quantity and timing are everything, and as current diagnostics can attest, knowing which stage a person is in is quite challenging.247248
Using calcium as a cofactor, tau released from cortical neurons is capable of activating AMPARs and thereby stimulating the neuronal hyper-activity. Without calcium, this phenomena is considerably less prominent.249 Nonetheless, antibodies to GluA3 AMPAR subunits as a result of autoimmunity also reduce their levels with or without tau’s involvement. This has been shown to lead to a loss of dendritic spine density along with increased levels of tau and neurons. This could be interpreted to mean that disruptions in immune function are of higher significance than the presence of tau, itself.
Just how prevalent these antibodies are in the general population with dementia is difficult to say, because the sample size has been so small. Nonetheless, given what I have been describing here, you may agree with me that GI disturbances and shifts in immune function may very well be at the root of dementia pathology.250 Others are clearly thinking along these lines and some scientists have even been arguing recently that autoimmunity is, in fact, one of the primary causes for dementia. One study, for example, demonstrated higher anti-nuclear antibodies in patients with frontotemporal dementia as compared to control subjects.251
Given that aberrations in glutamatergic transmission are also common in amyotrophic lateral sclerosis, it may be assumed that ALS has an autoimmune origin as well. I will say this: I have seen more cases than I would like to admit where there is a history of viral infection (e.g. EBV, HSV, etc.), with autoimmunity on its heals. In at least a couple of those cases, there has been a diagnosis of either ALS or Parkinson’s. When the doctors were challenged with the question of whether or not the neurodegenerative conditions could be related to the history of viral infection, they rapidly denied the correlation. In my mind, this simply confirms the continued disconnect in the medical community between immunology and neurology.252
Though I won’t go into detail now on the subject, I will say that correlations have also been made between excess serotonin in the prefrontal cortex and overactive glutamatergic transmission. I will let you draw your own conclusions there, but suffice it to say that enterochromaffin cells in the gut release serotonin (among other things) in the presence of inflammation and in models of gut permeability, this excess serotonin escapes into circulation. Any number of factors listed above could permit serotonin outside the blood-brain barrier into the brain and frontal / prefrontal cortex. Can we still say that the problem is exclusive to the brain?253254
At this time, there are no approved treatments for frontotemporal dementia, and no therapies have been able to demonstrate the ability to delay its progression. Memantine, an NMDA receptor antagonist, is frequently prescribed for FTD as well as Alzheimer’s,255, but randomized, placebo studies have failed to prove significant benefits with regards to disturbances in behavior.256257
In my mind, the most promising therapeutic options at this time involve targeting of autoimmune disorders. AMPAR modulation is quite messy and it can be difficult to gauge “which side” of the activation curve one is on. Diagnostics are improving in this area, but going to the root cause is a better long-term strategy. Currently, the only trials that have been performed in this area have focused on autoimmune encephalitis involving either NMDA or AMPA receptors. These trials must be extended to include neurodegenerative diseases.258
One viable therapeutic approach might involve scavenging of anti-GluA3 antibodies, but in my opinion, it would not be effective without first understanding the original triggers for autoimmunity that are at the root of those antibodies in the first place.
Regardless of which therapy is embraced, it must take into consideration not only neurotransmitter abnormalities but also genetic mutations, environmental factors, and immunological status. Without looking at the entire picture, therapies will continue to fail. In the meantime, we should continue to develop and improve existing technologies for the detection of glutamate abnormalities, including EEG and TMS-EEG.259260 Positive modulation of glutamatergic transmission could be useful for improving memory and learning, but it carries the risk of enhancing excitotoxicity and neuronal death.
The point I am driving toward here is quite simple: antagonizing glutamatergic receptors for the purpose of neuroprotection confers greater risk for memory impairment and cognitive decline. Among those in the biohacker community, I frequently see MCTs being combined with AMPA-receptor agonists such as the racetam family of nootropic “brain drugs”. Intuitively, this strategy may seem reasonable for keeping glutamatergic balance, especially if MCT consumption is at several tablespoons per day, but again, it is extremely difficult to know which side of that balance you are on. Both agonizing and inhibiting AMPARs carry significant risks. It is therefore my opinion that modulation of the AMPA system should generally be avoided unless there is a familial history of dementia or currently relevant symptoms.
Ironically, the very mechanism by which MCTs control seizure in models of epilepsy is the very means by which they can also induce cognitive decline. At the onset of Alzheimer’s disease, downscaling of AMPA receptors is now regarded as one of the main causative factors in cognitive impairment. In other words, whereas glutamate over-activation of AMPA receptors is in part responsible for neuronal excitation leading to seizure, an inability to adequately activate those same receptors due to downscaling is at the root of cognitive decline in Alzheimer’s.
In the early stages of dementia, synaptic disruption alone is enough to induce loss of memory, with or without damage to neurons.261262263 Amyloid is known to impair synaptic plasticity in brain regions such as the hippocampus,264, characterized by disruptions in long-term potentiation (LTP), the process by which memories are encoded.265
LTP impairment has become a sensitive marker in the early stages of Alzheimer’s due to its impairment well before amyloid plaques begin to accumulate.266267 The late phase of LTP, also called “expression” is the most vulnerable to impairment.268269270271 This expression phase absolutely depends on AMPA receptors and involves, in part, their recruitment to synaptic membranes.272
It has even been shown in studies that removal of AMPA receptors leads to a significant drop in excitatory transmission and the induction of “long-term depression”, or LTD.273274
A process of homeostatic synaptic scaling has been defined which describes the ability of the AMPA receptor network to increase or decrease receptor density to modulate total synaptic strength.275 This process, most fundamentally, involves the insertion or removal of AMPARs at post-synaptic terminals.276277278 As such, synaptic scaling is the primary mechanism by which synaptic strength is regulated in the process of learning.279
Without the ability to adapt AMPA receptor density, neurons lose their ability to properly engage LTP, and synaptic strength declines, resulting in the inability to both form and retrieve memories.280281 Therefore, anything that disturbs the homeostatic mechanisms that regulate AMPARs can potentially lead to significant cognitive impairment. Obviously, with its ability to inhibit AMPARs directly, decanoic / capric acid is a strong candidate for functional AMPA “impairment” in models of early onset dementia. Where do we draw the line between mitochondrial enhancement and AMPA network downscaling?
To be clear, in the early stages of Alzheimer’s, when symptoms are barely noticeable (if at all), a selective decrease in AMPARs has been noted. Any inhibition of those receptors whatsoever during that stage will not only enhance the physiological and cognitive consequences but also accelerate them. Once again, this is precisely what MCTs do in the brain in models of epilepsy.
AMPARs already decrease in number with age, without markers of dementia. In this sense, regardless of the presence of amyloid, we can presume that any inhibition of that system in older age could potentially produce symptoms of cognitive decline not unlike those seen in frank dementia. The only exception, of course, would be models of neuronal hyperexcitation such as seizure.282
Given that behavioral and physiological impairments frequently precede the deposition of amyloid plaques, many scientists now believe that soluble, neurotoxic amyloid is capable of inducing a reduction in AMPA receptor density. Beta-amyloid levels sharply rise in parallel with AMPA receptor downscaling, at an age when plaque levels are yet low.283284285 AMPAR downscaling is a vital process for protecting neurons from damage from either elevated levels of excitation or decreased inhibition.
The fact that this ability is gradually lost as dementia progresses clearly argues, in my mind at least, against the excessive use of dietary MCTs at any stage of dementia. Seeing how decanoic acid can accumulate, especially in the presence of C8 and that this accumulation is exquisitely carnitine-depletion sensitive – along with the other factors described that can effect MCT metabolism via ACADM – it should be blindingly obvious that things can get out of hand in the brain quite quickly, depending on a wide variety of conditions. Once again, make no mistake about it: these effects could be unnoticeable and go on for many years before significant symptoms appear.
The irony of MCT therapy for Alzheimer’s disease is striking. Regardless of the other beneficial effects in models of dementia listed above, the very system that is in decline as AD progresses is the very system that is inhibited, at increasing MCT concentrations. Currently, more forward-thinking proposals for AD treatment are considering strategies to maintain AMPA receptor activation and halt downscaling. 286287 Indeed, compounds are already appearing that have shown the capability to modulate AMPARs, and the results have been quite encouraging.288 That being said, I cannot imagine that inhibiting this system to any degree would be desirable in one that is trying to avoid dementia.
Calcium and excitotoxicity
Calcium plays a much bigger role in dementia pathology than many realize. Given that excess extracellular calcium can directly influence synaptic strength as a cofactor in AMPA receptor activation, it has long been a target for dementia therapeutics.
In Alzheimer’s, soluble forms of amyloid-beta are capable of provoking calcium influx through neuronal plasma membranes using calcium-conducting ion channels. Over time, this leads to excitotoxic neurodegeneration.289 For this reason, calcium channel blockers have been considered as a strategy to mitigate amyloid-induced neurodegeneration. In animal models, such compounds have been demonstrated to be neuroprotective. Even in cases of vascular dementia, calcium channel blockers are able to relax the cerebral vasculature.
Several clinical studies have investigated the effects of calcium channel blockers on dementia. Such studies primarily used the dihydropyridine Nimodipine, but the outcomes have been less than impressive.
Responses to electrical and chemical stimulation are primarily mediated by calcium. Keeping extracellular calcium levels balanced is vital to neuronal survival and function. As we get older, we gradually lose control of intracellular calcium concentration, due in part to neuronal membrane permeability, allowing more extracellular calcium into the cytosol.290 As I mentioned in my article on seafood consumption, membrane permeability is also exacerbated by excess dietary DHA and EPA. Glutamate, released at central synaptic sites, facilitates calcium’s entry at postsynaptic sites via NMDA receptors and indirectly through L-type calcium channels. An overabundance of glutamate can lead to excessive calcium loads at those sites, provoking a signaling cascade that eventually results in the death of neurons. This process has been coined the name “excitotoxicity” and is established as the underlying pathology for most neurodegenerative disorders, including Alzheimer’s.291292293294
By blocking calcium influx in both synaptic terminals as well as postsynaptic sites, excitotoxicity may be mitigated. The primary problem with calcium antagonists is that they are frequently non-selective and can have dangerous side effects, especially with regards to the cardiovascular system. Further, these antagonists have a very low brain to plasma ratio, given they are poorly transported through the blood-brain barrier. Compounds such as TROX-1 work around these limitations and seem to have fairly optimal selectivity. Nonetheless, I believe the problem to be less about what calcium is doing and more about why levels may be high to begin with. The answer to this question is beyond the scope of this article, but it includes a number of factors including dietary calcium intake, vitamin D and K status, as well as any compound that could potentially affect calcium homeostasis.295 One such compound is caprylic acid.
An interesting study investigating sensitivity to odors demonstrated that caprylic, i.e. octanoic acid, provokes increased release of calcium ions into the cytosol of cells. This is an important finding. Though this particular study doesn’t discuss calcium dynamics in other parts of the body, we have specific evidence that octanoic acid is capable of directly influencing cellular calcium influx. This has wide-reaching implications for the dementia pathology. A conclusion may be drawn that octanoic acid in the brain could potentially exacerbate calcium-channel mediated excitation of glutamatergic neurons. Indeed, this effect may very well be one of the ways in which biohacker anecdotes of “improved cognitive function” have appeared — via higher glutamatergic signaling. This should also lead us to believe that lower levels of MCTs in the brain could be stimulatory, especially if the dominant MCT is octanoic acid. These points only further limit our ability to determine the safety of MCT consumption in each individual case.296
Caprylic acid is not the only guilty party with regards to calcium, however. Yet other studies exist which clearly demonstrate the ability of capric (i.e. decanoic) acid to alter calcium ion homeostasis in cells, especially in the presence of compromised metabolism of medium-chain fatty acids.297
Calcium-Induced Mitochondrial Death
As we’ve already discussed, excess octanoic and decanoic acids in the brain behave as metabolic inhibitors of mitochondrial oxidative phosphorylation. This is especially true for decanoic acid which is not well metabolized, due to the absence of CPT1 expression in the brain. What I haven’t mentioned yet is that decanoic acid, in sufficient amounts, also decreases mitochondrial membrane potential and calcium ion retention capacity in brains with excess calcium.
These problems can go undetected for a lifetime and appear at any age in the event of prolonged fasting, chronic infection, or other types of metabolic stress.298299 We’ve talked about how L-carnitine may be used to contain this problem, but it has not proven capable of preventing neurological symptoms associated with aberrations in calcium signaling.300301
Excess decanoic acid decreases membrane potential in the mitochondria of the brain, and this effect is amplified in the presence of calcium. Further, excess MCTs in the brain also have the capability of increasing neuronal malondialdehyde levels, resulting from lipid peroxidation. This induces oxidative stress on neurons, leading to the formation of mitochondrial permeability transition pores (mPTP or MPTP) in the inner membrane.302303304305 Opening of mPTPs increase the permeability of mitochondrial membranes to molecules of less than 1500 daltons in molecular mass. This can result in swelling and, eventually, apoptosis (i.e. cell death). Decanoic acid further decreases mitochondrial capacity for calcium retention, thereby leading to a loss of control over calcium concentrations inside cells.306307308309310
For the reasons stated above, higher intracellular calcium in neurons results in hyperactivation of NMDA and AMPA receptors and, eventually, neuronal death. Given that the levels of C10 that must accumulate for AMPA receptor inhibition are quite high, it can be presumed that the excitatory effect is seen primarily at lower levels – in other words, average levels of MCT consumption. The rate of which the process unfolds is difficult to ascertain until there are apparent symptoms. Only in the case of full-blown MCAD disorder would issues materialize quickly and tangibly. Again, the severity of risk depends on the level of metabolic disruption from environmental stress. Infections or autoimmune responses can only add oil to this fire.
MCTs induce nitrosative stress
One of the other ways that decanoic acid can induce brain damage is via its inhibition of alcohol dehydrogenase (ADH3). ADH is responsible for reducing S-Nitroglutathione (GSNO), which plays a major role in nitric oxide (NO) signaling and is a source of bioavailable NO.311 GSNO is reduced by ADH3 to S-hydroxylaminoglutathione which, in the presence of reduced glutathione (GSH), forms oxidized glutathione (GSSG). Both GSH and GSSH inhibit glutamatergic signaling by binding to NMDA and AMPA receptors.312
By inhibiting GSNO reduction to GSH and GSSG, decanoic acid can potentially increase neuronal nitric oxide formation. This leads to increased neuronal nitrosative stress, overexcitation and neurodegeneration. Because ADH3 also plays a role in formaldehyde detoxification, this inhibition can further lead to formaldehyde accumulation in certain contexts.313 Not surprisingly, formaldehyde interferes with DNA-methyltransferase function, which is critical for both the formation of recent memory and the maintenance of remote memory.314 As a matter of fact, measurement of formaldehyde in overnight fasting urine has be been proposed as a noninvasive method for evaluation of dementia risk.315
In healthy brains, a certain amount of nitric oxide generated by SNOs is required for proper regulation of synaptic plasticity and neuronal survival.316 Dementia pathology, however, demonstrates elevated nitric oxide levels, where NO is implicated in both nitrosative stress and resulting neuro- inflammation.
To bring all these concepts together, let’s consider that nitric oxide synthase (NOS1) is tethered to glutamatergic neurons. NOS1 is activated by NMDA receptor calcium influx, followed by SNO-mediated modulation.317 When NMDARs (and AMPARs) are overactivated, there is a parallel overproduction of nitric oxide and “nitrosative stress” induced neuronal damage.
SNOs such as GSNO have been shown to be elevated in the hippocampus and cortex during the early stages of dementia, directly preceding the appearance of behavioral symptoms. In fact, excess SNO activity is believed to impact amyloid processing and clearance, increasing the potential for amyloid plaque deposition. Therefore, by inhibiting enzymatic degradation of GSNO, decanoic acid further contributes to SNO-protein mediated induction of nitrosative stress, inflammation, and accelerated progression of dementia, especially in the presence of the other factors I have been describing above.
These problems are compounded if there are any other sources of oxidative stress throughout the body. Over time, such stress will deplete glutathione stores, thereby further decreasing available GSH for the beneficial conversion of S-hydroxylaminoglutathione to oxidized glutathione (GSSG). This results in a net loss of glutamatergic inhibition, raising the risk for neurodegeneration.
Unfortunately, the glutathione story does not end there. Given that reduced glutathione, itself, has been shown to bind to NMDA and AMPAR receptors and inhibit excitotoxicity, GSH depletion has wide-ranging effects on synaptic transmission plasticity. In fact, a 40% decrease in brain glutathione level is sufficient to cause increased excitability in neurons.318
If you search the scientific literature, you will find that NMDA receptor hypofunction has been associated with glutathione deficit. This, in part, is due to the excessive oxidative stress induced by neuronal overexcitation. In other words, GSH deficit results in excessive oxidation of extracellular redox-sensitive sites on NMDA and AMPA receptors, effectively blunting their proper function.
Again, overconsumption of MCTs, especially decanoic / capric acid, can powerfully inhibit proper reduction of S-nitrosated glutathione, resulting in higher potential for nitrosative stress, neuroinflammation, and neuronal damage. Only at extremely high levels of consumption would we see any AMPA-inhibitory effect. This problem is compounded by the deficit in reduced glutathione that occurs when there are other stress reactions going on throughout the body, such as those seen in autoimmunity or inflammatory disorders, in general. If you believe that taking liposomal glutathione orally could potentially offset this problem, think again. By taking exogenous glutathione, you are effectively increasing the reduced glutathione pool, which may be S-nitrosated to GSNO, especially if there is any degree of pre-existing neuroinflammation. This is yet one more way in which MCTs are capable of single-handedly inducing changes, in this case in the brain, that create the perfect storm for dementia pathology.
MCTs reduce melatonin secretion
One of the other patterns I have noticed in clients that regularly include MCT oil in their diet is a low or severely depressed salivary melatonin level in the morning, combined with suboptimal circadian rhythm. My first impression was that they could either be low on precursors for melatonin such as tryptophan or serotonin or, possibly, dealing with some form of chronic stress that was raising cortisol levels and disrupting melatonin production. It wasn’t until I discovered that glutamatergic receptors are also found on pinealocytes – and actually play a role in melatonin secretion – that the pieces started to come together.319
Pinealocytes synthesize melatonin, which is not only a powerful antioxidant but also a biologic modulator of mood, reproduction, sexual behavior and, most importantly, sleep. In fact, melatonin’s regulation of the sleep-wake cycle is well known and melatonin supplements can be found nearly in every supermarket, drugstore, or pharmacy.320321322323324325326
Not a week goes by that I’m not encountering someone that has moderate to severe sleep disturbance and insomnia. The majority of them are low melatonin secretors (for a wide variety of reasons). Nearly all of them consume MCTs, either in the past or currently, and all of them are on a low-carb, high-fat diets.327 Many things can interfere with melatonin secretion, not the least of which is stress from any source, hypoxia, or even just taking exogenous melatonin itself.328329
In the late 90s, we discovered that glutamate mediates the secretory activity of the pineal gland.330331 The NMDA and AMPA glutamate receptors have been shown to exist in pinealocytes, especially AMPA subunit GluR1.332 In fact, it is thought that AMPA GluR2/3 receptors even play a role in neuronal differentiation, including that of pinealocytes, further modulating their function during pineal gland development.333
The AMPA receptor GluR1 is thought to directly participate in the regulation of melatonin secretion.334 This may be one of the factors, aside from neuronal oxidative stress, that plays into circadian rhythm disruptions and insomnia seen in Alzheimer’s disease. Because pineal melatonin secretion depends on proper glutamatergic signaling, disruptions in that cascade could lead to lower melatonin levels.
Therefore, it is also not out of the question that in the case of decanoic acid overconsumption, especially in parallel to octanoic acid, AMPARs of pinealocytes could be inhibited in a similar manner to that seen in epilepsy therapy, thereby leading to hyposecretion. This hypothesis is not well supported at this time, and I encourage any scientists reviewing this material to organize a study that would elucidate it further. As for my opinion, I cannot deny the low salivary melatonin seen in my clients that observe MCT-loaded ketogenic diets. Is this the result of C10 accumulation?
MCTs may disrupt the circadian clock
Another possibility for how decanoic acid may disrupt sleep is via its inhibition of the alcohol dehydrogenase that plays, in part, a role in retinol metabolism to retinoic acid. Inhibition of this reaction to any degree will reduce retinoic acid levels. Retinoic acid plays a major role in circadian rhythm by regulating hippocampal gene expression for the circadian clock, delta oscillations in slow-wave sleep via RAR-beta, and vasopressin neuronal activity in the suprachiasmatic nucleus. Unless you are consuming beta-Carotene (and converting it adequately to retinoic acid), dietary transformation of dietary Vitamin A by alcohol and retinol dehydrogenases could potentially be limited by excess MCT consumption.335336
MCTs Promote Inflammation
 We have already considered the immune-modulating potential of MCTs and their ability to increase allergic sensitization. I want to turn our attention now to a deeper discussion of the inflammatory process, itself. Chronic, low-grade, systemic inflammation is a condition characterized by the constant presence of inflammatory molecules in circulation such as TNF alpha, IL1B, IL6, CRP, etc.
We have already considered the immune-modulating potential of MCTs and their ability to increase allergic sensitization. I want to turn our attention now to a deeper discussion of the inflammatory process, itself. Chronic, low-grade, systemic inflammation is a condition characterized by the constant presence of inflammatory molecules in circulation such as TNF alpha, IL1B, IL6, CRP, etc.
Adipose tissue, itself, is capable of producing and secreting many of these molecules, which is why obesity is almost always correlated with one or more inflammatory diseases. The more systemic inflammation is, the higher the potential there is for insulin resistance, high blood sugar, endothelial dysfunction, and ultimately stroke and dementia.
Inflammasome induction
Octanoic acid, on its own, is highly pro-inflammatory. It has been shown to modulate multiple genes associated with inflammation in skin, lungs, gut, and brain, including CASP1, IL1B, CD86, and ICAM-1. ICAM-1, for example, is expressed primarily in endothelial cells and cells of the immune system. It is well known for being used by the common cold (i.e. rhinovirus) as a receptor that facilitates access to the respiratory epithelium.
ICAM-1, also known as CD54, is frequently found in sites of inflammation and can be directly induced by common inflammatory cytokines such as IL-1 and TNF-alpha. Upon activation, leukocytes bind to endothelial cells via ICAM-1 before transmigrating into tissues. ICAM-1 further plays a major role in allergic reactions and has even been shown to promote hypersensitivity reactions by recruiting mast cells. ICAM-1 expression is acutely activated in dendritic cells when exposed to octanoic acid.337
This reaction is very similar to dendritic cell response to lipopolysaccharide, so it would be expected that octanoic acid, in the gut for example, would increase ICAM-1 activation and inflammatory cytokine activity even more strongly in the presence of gram-negative bacteria.
N-Acetyl Cysteine (NAC) has shown the capability to suppress ICAM-1 expression, but it is unable to do so in the presence of octanoic acid. This should give you some idea of how profoundly pro-inflammatory octanoic acid may be, especially in the presence of antigens.
Leukocyte inhibition
Another study demonstrated that MCTs, in the presence of existing infection or other biological stress, directly stimulate immune cells, increasing production of reactive oxygen species (ROS) and expression of adhesion and granulation markers, thereby inflicting deleterious effects on critical leukocyte functions such as cell migration and bacterial killing.338339340341
Such effects are not prominent in healthy individuals, but studies have not followed MCT consumers over longer periods of time, when the effects of chronic consumption become more apparent. For example, one might be entirely asymptomatic on a high MCT diet for 20 or 30 years or more, only to find sudden onset dementia via the mechanisms described above.342
Gastric inflammation and ulcers
Both octanoic and decanoic acids have additional effects on the inflammatory profile by inhibiting cyclooxygenase-I (COX-1)343 COX-1 is an enzyme that generates prostaglandins from arachidonic acid. Its activity is regulated by cytokines and growth factors.
Inhibition of this synthase is not desirable, given that it is a regulator of angiogenesis in endothelial cells and plays a role in cell signaling. One of COX-1’s most important functions, in fact, is its promotion of the natural mucus lining that protects the inner stomach. It thereby limits acid secretion and pepsin content, reducing risk for gastritis.344 Inhibition of COX-1 has been shown, in some studies, to lead to gastric ulceration. For this reason, many of the anti-inflammatory compounds that work on COX selectively target COX-II. As a matter of fact, octanoic acid shows absolutely no COX-II inhibitory activity.
MCTs Cause GI Disease
 Perhaps the most troubling effect of MCTs are their ability to affect the localization of claudin which plays a crucial role in tight-junction integrity in the gut.345 The sodium salt of decanoic acid (sodium caprate), for example, is a well-known enhancer of tight-junction permeability.
Perhaps the most troubling effect of MCTs are their ability to affect the localization of claudin which plays a crucial role in tight-junction integrity in the gut.345 The sodium salt of decanoic acid (sodium caprate), for example, is a well-known enhancer of tight-junction permeability.
When decanoic acid comes in contact with tight-junctions, it changes the distribution of actin filaments and claudin proteins, specifically claudin-1 (CLDN1). Ordinarily, only ions, water, and water-soluble molecules are allowed through the tight-junctions of the gut. These junctions are comprised of occludin, claudin, and junctional adhesion molecule (JAM).346
Though occludin is important for organizing the structure of the tight junctions, it does not play a role in their assembly. Rather, it is the claudin family that is primarily responsible as the gate-keeper of the gut,347 and they can be regulated by various stimuli that include foods, drugs, and chemicals. In particular, decanoic acid has been shown to increase the passage of otherwise prohibited molecules across the tight junctions by contracting actin filaments.348349
To be clear, the primary job of tight junctions is to ensure that allergens, toxins, pathogens, and other unwanted molecules do not make their way into circulation. Decanoic acid increases paracellular transport of high molecular mass compounds across the tight junctions, with transport efficiency actually increasing with larger molecules. Sodium salts of these compounds are even more efficient in this respect.
In multiple experiments, tight junctions treated with C10 were shown to contain fragmented claudin-1 proteins with a significant decrease in overall distribution of occludin, JAM-1 and zonulin. One of the markers of membrane damage in cells is lactate dehydrogenase (LDH), and LDH levels have also been seen to increase when exposed to C10 MCTs.350 In other words, in order to achieve higher paracellular transport across tight junctions, these MCTs induce damage to the junction membranes.
It has been demonstrated that in order to enhance paracellular transport across tight junctions, an increase in both the radius and number of tight junction pathways must occur.351 This creates multiple pores in the gut barrier, and C10 MCTs have proven to create larger pores than other substances used for this purpose. One study even reported the pore radius in the tight junction pathway increasing from 8 to 20 angstroms in the colon on contact with capric acid.352
Keep in mind that the larger the pore size, the wider array of molecules, including microbial endotoxins, can escape the gut lumen and enter the bloodstream. MCT oil directly facilitates this process, and more effectively than nearly any other compound.
Severe tight junction disruption
One of the reasons scientists were studying the ability of MCTs to increase gut permeability was for its potential application in the enhancement of pharmaceutical absorption. Many hydrophilic compounds, such as peptide- and protein-based medications, are unable to penetrate cell membranes, thereby limiting their ability to achieve the desired effects. For quite some time now, pharmaceutical companies have been looking into strategies that involve improved absorption of hydrophilic macromolecular drugs with the co-administration of absorption-enhancing agents.353
In fact, one of the first applications of sodium caprate was for the enhanced absorption of rectal ampicillin suppositories. These days, the same effects are being achieved with pure MCT oil.354 Another example of how MCTs are being used is with their application in the administration of oral insulin. Orally administered insulin has very low bioavailability – not so when combined with MCTs. Are you beginning to see the larger picture here?355356
Nature has always understood the value of medium-chain triglycerides for the enhancement of absorption. Why else would capric acid be present in human breast milk?357 Tight junctions are located just about everywhere there are epithelial tissues. This includes the intestine, skin, mammary epithelium, pleura of the lungs, kidney, and – of course – the brain capillary endothelium.358359360361362363
The glue that holds tight junction proteins together is known as tricellulin,364 which is located at the meeting point of three cells where three bicellular tight junction strands converge.365 Without tricellulin, the entire tight junction network would collapse.366 As a matter of fact, overexpression of tricellulin has proven to make the tight junction barrier virtually impermeable.367 All that is needed to allow larger molecules such as ampicillin or insulin through the gut barrier is to incubate them in capric acid / C10.368369 In addition to the effects I have noted above, capric acid has also proven to have another rather disturbing capability: the ability to decrease tricellulin directly.
Though capric acid’s effects are thought to be transient, the redistribution of tight junction proteins, decrease in tricellulin, and creation of wide-radius pores is not an insignificant issue. With repeated, chronic use of MCTs, it should be quite obvious that in addition to provoking allergies to foods and other substances, they will also open the tight junctions (transient or otherwise) and allow passage of some potentially very undesirable things. Gut permeability is hallmarked by increased circulating endotoxin and systemic inflammation. MCTs produce this condition, regardless of whether or not symptoms are present.
To add insult to injury, MCTs also lyse bacterial membranes in the same manner they degrade tight junctions (in the process of exerting “antimicrobial” actions). This further elevates endotoxin load. I want to point out here that an antimicrobial compound that also opens tight junctions is certain flirtation with systemic inflammation, if not sepsis. While bacterial killing is useful, together with increased gut permeability, it is worse than no killing at all.
Colorectal cancer metastasis
It should be further concerning to find that capric acid is produced endogenously by colorectal tumors and has started to get attention as an early diagnostic biomarker for colorectal cancer.
Recent statistics show that colorectal cancer (CRC) is the second leading type of cancer in women and third leading type in men.370 Having a history of polyps, inflammatory bowel disease, ulcerative colitis, or Crohn’s disease all put a person at higher risk for developing this deadly cancer. As mentioned above, carnitine deficiency is thought to be behind the ulcerative colitis pathology, in particular, and this can be induced by MCT overconsumption, alone. Given that colitis is a risk factor for CRC, MCTs effects can easily be extended to cancer promotion as well. Unfortunately, that is not the whole story for MCT / CRC correlations.
A new branch of metabolomics called “lipidomics” has been looking critically at the metabolic state of cells and tissues, in the context of various diseases, to understand what role lipids play in their development.371372373374 Diets have a profound effect on microbial populations of the gut, though lipid profiles can change drastically from nothing more than shifts in bacterial fermentation or even fatty acid synthesis directly in cancer tissues.375
It has long been established that overconsumption of long-chain fatty acids reliably increases risk for colorectal cancer, but much controversy exists over how it develops. As I have described thus far, fatty acids clearly play a wide gamut of roles in cellular metabolism. Polyunsaturates such as linoleic and alpha-linolenic acid, for example, are lower in colorectal cell membranes, whereas arachidonic levels are increased.376 If we look at the plasma of colorectal cancer patients, indeed, LA and ALA are low there as well.377378
MCTs, on the other hand, are not so easy to measure. Most make their way directly to the liver via portal circulation and are beta-oxidized in mitochondria and / or converted to ketones. Liver enzymes responsible for lipid release into serum after feeding are more efficient with fatty acids of 14-carbons or more. MCTs, however, don’t tend to be reflected very well in blood serum unless one is overconsuming them (as on an MCT ketogenic diet).379
Lipids are synthesized in cancer tissues via the activation of oncogenes.380 In fact, increased fatty acid synthesis is part and parcel to cancer development. This is especially true in the case of colorectal cancer tissues, which show overexpression of fatty acid synthase (FAS). Indeed, high FAS expression is correlated with a poor prognosis in colorectal cancer.381 Nonetheless, FAS is not very useful as a screening tool because it requires tumor resection. Therefore, scientists have turned their attention to fatty acid levels in the blood as an early screening strategy.
With regard to colorectal cancer cells, statistically significant elevations of both octanoic and decanoic acids have been shown. At least two studies I am aware of have correlated MCT overconsumption with increased risk for colorectal cancer, while other studies have negated this association.382383384 Apparently, negative correlations have been primarily associated with dodecanoic acid, which is abundant in coconut and palm oils. Such studies did not look at the C8 and C10s.
That being said, I find it rather alarming that colorectal cancer patients have been accurately distinguished from healthy people with 82% and 87.7% probabilities using only C8 and C10 fatty acids, respectively, as diagnostic markers. In my opinion, it is no coincidence that C10 levels correlate more specifically to colorectal cancer progression, considering what I have already described regarding its effects on adhesion molecules and tight junctions. From the perspective of CRC pathology, it makes sound biological sense that tumors would directly leverage MCTs to improve viability, translocation and transmigration (i.e. metastases).
Unfortunately, colorectal cancer’s high capability for metastasis has been clearly proven to involve the fatty acid oxidation pathway, and it should not surprise you that CPT1A is upregulated in CRC cells.385 As I have mentioned, one of the reasons that decanoic acid accumulates in the brain is due to the absence of an active brain-centric CPT1 isoform. Knowing that colorectal cells invoke FAS to produce MCTs in parallel with CPT1 upregulation, how do you now feel about MCT consumption, in general?
MCTs Disrupt Liver Detoxification
 Clearly, I could conclude this article now with only the information I have shared thus far. The truth of the matter is, however, that my research on the subject has revealed other areas which are equally as important for the purposes of thoroughly and completely quantifying the risks of MCTs.
Clearly, I could conclude this article now with only the information I have shared thus far. The truth of the matter is, however, that my research on the subject has revealed other areas which are equally as important for the purposes of thoroughly and completely quantifying the risks of MCTs.
The time has come to take this poison off the shelves, and I need for you to have all the facts to come to this same conclusion. Without such a complete perspective, there is a possibility that it may continue to be a popular dietary item, and my conscience would simply not permit that to happen. So we must continue.
You might be wondering at this point – what exactly happens to MCTs if they are not properly utilized or there has been an accumulation due to overconsumption or other metabolic block? The answer lies in the liver, where excess MCTs and their metabolites are glucuronidated in preparation for their excretion.
Impaired Glucuronidation
Glucuronidation is a Phase II liver-mediated detoxification process that involves the conjugation of lipophilic compounds, some potentially toxic, with glucuronic acid to faciliate their expedient excretion. Glucuronic acid, or “glucuronate”, is actually a sugar acid derived from glucose (in the pathway for pentose-glucuronate conversions). While I won’t go into detail about the genetic issues some people have with making their own glucuronic acid, suffice it to say glucuronic acid insufficiency is a larger problem, in my opinion, that the medical establishment has recognized, especially in a modern world full of increasingly large volumes of toxin.
Not only is glucuronate used for conjugation in the liver, but it is also a building block for proteoglycans and glycoglycerolipids such as heparin, chondroitin sulfate, dermatan sulfate, keratan sulfate, hyaluronic acid, etc. so if you have depleted your glucuronate supply for any reason, you are also likely to suffer deficits in these other macromolecules.
UDP-glucuronyltransferases (UDPGTs) are the enzymes primarily responsible for attaching glucuronate molecules to their target compounds. Though glucuronidation mainly occurs in the liver, UDPGTs have also been identified in almost every major organ of the body, including the intestine, kidneys, brain, adrenals, and even the prostate.
The conjugated products of glucuronidation are known as beta-D-glucuronides. Once these glucuronides have been formed, they are eliminated from the body via urine or stool much more quickly than in unconjugated form. In order that they be incorporated into stool, they must be mixed with bile, so bile secretion disorders will also play into the dynamic here.
The problem with glucuronidation is that it is rate-limited by glucuronic acid supply, and it is possible to exhaust that supply when combining drugs or substances whose secretion is primarily or entirely dependent on that process. Even though there are secondary detox routes in the event glucuronidation becomes overwhelmed, they are not as fast or efficient, and there is a tendency for accumulation of substrates. The most common case of this issue involves prolonged drug effects due to delayed clearance. In more severe crises, substrate accumulation can be potentially lethal. Such is the case with MCAD deficiency, when the ability to remove MCT metabolic products becomes impaired.
Not long ago, it was established that MCTs use glucuronidation as a primary excretion route. Therefore, any other substrate that uses this process can potentially compete for priority. Such competitors include estrogens and their metabolites, testosterone, androsterone, etiocholanolone, bilirubin, and retinoic acid. On top of these substrates, alcohol, morphine, Tylenol, NSAIDs, and benzodiazapines are all capable of contributing to glucuronic acid depletion. The question really lies in which substrate “gets there first”.
To make the issue more complicated, intestinal microflora are capable of producing beta-glucuronidase, which hydrolyzes glucuronides, thereby releasing their substrates back into circulation. For this reason, individuals with high gut beta-glucuronidase levels also tend to have sex hormone imbalances. I will leave it to your imagination to surmise what happens when these same people adopt an MCT-rich ketogenic diet.
I can cite at least two cases I have observed that involved a high-MCT consuming individual that developed yellow-tinted skin due to bilirubin accumulation. A short course of Calcium-D-Glucarate along with MCT avoidance was all that was required to resolve the problem, though I have seen MCT exclusion, alone, resolve such imbalances. This furthers my opinion that excess MCT can also interfere with bilirubin excretion.
Unfortunately, there is another twist to this story, and it once again involves carnitine. If we turn to the literature on MCAD, we see prominent excretion of octanoylglucuronide (a metabolite of caprylic acid). It’s important to note that we would see little to no metabolites in conditions where glucuronic acid reserves were low.
Considerable excretion of saturated, unsaturated and hydroxylated medium-chain fatty acids has also been seen, along with several conjugates such as suberylglycine, hexanovlglycine and octanoylcarnitine.386387 Of note, these urinary profiles are accompanied by a parallel low level of carnitine in both urine and plasma. Earlier on, glucuronidation of medium-chain fatty acids was associated primarily with valproic acid in the context of epilepsy treatment.388 That being said, if octanoic plasma levels go high enough, they can also provoke hepatic encephalopathy.389
The problem here is that when MCT serum levels go too high, carnitine conjugation is used as a secondary excretion mechanism. So not only is carnitine depletion exacerbating proper MCT metabolism, it is also being pulled into the elimination process as well, thereby lowering carnitine stores further and increasing potential for MCT hydroxylation. This latter process, in particular, is known for its production of metabolites that disrupt the urea cycle and increase ammonia levels. In this way, carnitine deficiency induces neurotoxic ammonia accumulation390
This situation can largely be avoided if there are sufficient glucuronic acid stores, but seeing as C8 and C10 prefer glucuronidation, carnitine conjugation is eventually inevitable, the longer MCTs and their metabolites accumulate. As I will describe below, glucuronidation of octanoic acid is particularly important due to its direct ability to induce gluconeogenesis. This can lead to reactive hypoglycemia in those so prone – such as those on a ketogenic diet with suboptimal insulin signaling or for individuals with insulin resistance.391
Polyphenol consumption adds to the problem
There is another factor that can play into glucuronidation imbalances, and it is probably the least likely suspect of all: polyphenols. Also known as polyhydroxyphenols, this family of chemicals are present in a large array of foods, most generally considered to be health-promoting. While the majority of polyphenols are known to be absorbed in the small intestine, those that escape absorption make their way into the colon, where they can undergo several structural modifications. One such modification is performed by colonic microflora, which hydrolyze glucuronides into aglycones and degrade them into simple phenolic acids.392393
In fact, this is precisely how the soy isoflavone daidzein is transformed into equol (which has far-reaching hormonal effects).394395396397 Once polyphenols become aglycones, they are methylated, sulfated, or glucuronidated in the liver.398399 For example, COMT catalyzes methylation of polyphenols such as quercetin, catechin, caffeic acid, and cyanidin, whereas the sulfotransferases (SULTs) and UDPGTs handle polyphenols found in common foods such as onions, artichokes, and kale.400401
For those individuals that observe diets rich in phenols or polyphenols, due to their affinity for UDPGTs, they can also potentially deplete the glucuronic acid store. In humans, glucuronides predominate for phenols and phenol precursors (such as benzene). In other words, diets rich in berries, onions, artichokes, potatoes, cabbage, kale, leeks, celery, broccoli, or parsley could easily lower your glucuronic acid store, thereby increasing the risk of accumulation for any one of the substrates I have mentioned above, not the least of which are MCTs and their metabolites.
The Final Insult: Fire in the Brain
 Last, but certainly not least, I would like to talk about MCT interactions with the endocrine system, specifically glucocorticoids. As I have mentioned above, one of the reasons that MCT-laden diets have been adopted for weight loss is because they are much less likely to promote adipocyte differentiation, due to their absorption directly into portal circulation. Nonetheless, caprylic acid has shown the ability to produce more fat cells in the presence of dexamethasone.
Last, but certainly not least, I would like to talk about MCT interactions with the endocrine system, specifically glucocorticoids. As I have mentioned above, one of the reasons that MCT-laden diets have been adopted for weight loss is because they are much less likely to promote adipocyte differentiation, due to their absorption directly into portal circulation. Nonetheless, caprylic acid has shown the ability to produce more fat cells in the presence of dexamethasone.
Dexamethasone is a corticosteroid medication with properties similar to cortisol. In one experiment, consumption of octanoic acid alone for 9 days had no effect on adipogenesis (i.e. the production of new fat cells). By adding dexamethasone, however, for an initial period of time, lipid droplets were already appearing at day 3. By the time day 9 was reached, lipid accumulation was present in nearly 50% of all cells.402
The higher the dose of octanoic acid, the more prominent the lipid accumulation. It has been hypothesized that corticosteroids may facilitate the storage of octanoic acid and its metabolites in cellular triglycerides, partially via the p38-MAPK pathway – a pathway which has been shown to govern adipocyte differentiation. In fact, octanoic acid stimulation of adipogenesis is specifically mediated by that pathway.
As it turns out, many stimuli can trigger the activation of p38 MAPK, including, but not limited to, ultraviolet light, radiation, heat shock, high osmotic stress, and most importantly – inflammation. Given MCTs ability to alter cell structure via alterations at membranes, it is highly possible that they could create a “stress condition” that invokes the p38 MAPK pathway. Unfortunately, octanoic acid is capable of increasing cortisol all on its own, with or without a stress response, and it does this via chronic activation of gluconeogenesis.
Caprylic acid induces chronic gluconeogenesis
Gluconeogenesis is a metabolic pathway that generates glucose when there is either insufficient carbohydrate in the diet, during fasting, or as a result of intense exercise. It catabolizes proteins to form glucogenic amino acids and breaks down triglycerides, both of which may be used for glucose production. Many individuals on a low carb, high fat diet or even those that replace breakfast with MCT-infused coffee for the purpose of “intermittent fasting”, are often driving blood glucose levels low enough to chronically invoke gluconeogenesis.
Generally, the liver and muscles will first be tapped by liberating glucose from glycogen. Those that engage in resistance training tend to have higher muscle mass and, therefore, higher glycogen stores. Individuals with low lean body mass, however, are relying exclusively on liver glycogen and, over time, because of carbohydrate restriction, can enter a phase of perpetual gluconeogenesis. In other words, if someone is consistently in ketosis, whether it be perpetual or cyclical, it is very likely we will see gluconeogenesis in parallel. The problem with this scenario, contrary to popular belief, is that while there may be better insulin sensitivity in the beginning, constantly invoking gluconeogenesis can (and does) lead to progressively higher blood sugar levels. In fact, the anti- gluconeogenesis diabetic drug, Metformin, lowers blood sugar levels by inhibiting, while also stimulating glucose uptake in cells.403 MCTs, in this respect, are acting in direct opposition to Metformin. Can you guess why Metformin is a popular off-label anti-aging drug for keto enthusiasts?
The problem here, as you might have guessed, is the means by which gluconeogenesis is invoked. While it can be stimulated by glucagon, growth hormone, adrenaline, and cortisol, chronic activation tends to prefer cortisol. This happens because of consistent and increasingly more severe drops in blood sugar (as glycogen stores are depleted). The lower blood sugar goes, the greater the emergency “stress response” is generated. In the early stages, cortisol is mobilized to provoke gluconeogenesis but, over time, the HPA-axis (hypothalamic-pituitary-adrenals) becomes less efficient and asynchronous, leading to more regular release of adrenaline. Again, complications are less frequent in athletic people that engage in resistance training, but I have seen this problem even in that subset, especially those that overtrain, such as the “CrossFit” group.
In almost all people that have embraced the ill advice for intermittent fasting with coffee (particularly coffee saturated with MCT oil), sooner or later, chronically high cortisol shows up and, with enough time, the tell-tale “love handles” around the waste.404 This is, in part, due to cortisol’s own ability to promote fat storage, but as I have already mentioned above, cortisol in the presence of octanoic acid will exponentially increase fat storage. And this all happens because of the disruption in glucose homeostasis that occurs when people consume MCTs without minding their glycogen status.
Further, when octanoic acid and adrenaline (or adrenaline-like compounds such as phenylephrine) are seen together, they produce additive effects on gluconeogenesis.405 In fact, octanoic acid has been shown to stimulate this pathway directly via the conversion of pyruvate – and this happens, regardless of ketone levels or rate of fatty-acid induced energy production (via beta-oxidation). In other words, octanoic acid appears to invoke gluconeogenesis regardless of blood glucose status, and this effect is only compounded by additional cortisol (or adrenaline) during stress responses such as fasting or intense exercise / training.
For this reason, we frequently see people in ketosis or a fasted state that also have blood sugar readings above 100 mg/dL. For the ordinary human, this is considered to be “pre-diabetic”. In fact, the optimal level in anti-aging groups has recently been set at 80-85. Most of the keto enthusiasts claim this is perfectly normal and even expected, and is not a sign of insulin resistance. I can only partially agree, because sustaining blood glucose levels above 100 mg/dL for any length of time will promote cardiovascular inflammation, regardless of other health markers. The literature has been quite clear on this point.
And – where there is high glucose in the blood, there is also high glucose in the brain. Given enough time, this leads to insulin resistance directly in the brain.
FoxO: Jekyll becomes Hyde
The correlation between brain insulin resistance and Alzheimer’s disease is well known. Epidemiological studies have shown increased risk for dementia in those individuals with diabetes mellitus (and vice versa).
As we have already established, oxidative stress is one of the primary drivers for the neurodegeneration seen in dementia, and not only does it lead to insulin resistance (by blunting or even blocking insulin binding to receptors), but insulin resistance itself provokes higher blood glucose levels – and glucose promotes inflammation and further oxidative stress. Once it has begun, it is a self-perpetuating cycle and in the majority of cases, this cycle begins slowly and unnoticeably.
One of the primary transcription factors responsible for liver gluconeogenesis as well as the cellular response to oxidative stress is FoxO (forkhead box class O). FoxO controls transcription of gluconeogenic enzymes and in the absence of insulin, it invokes hepatic glucose production. As insulin resistance progresses, the liver gradually loses control over FoxO and there is less inhibition of its transcriptional potential. In this way, upregulated FoxO activity promotes high blood sugar and, eventually, a runaway train for oxidative stress.
As nature would have it, FoxO seems to understand that its gluconeogenic activity can incur collateral damage, so we also see this protein controlling oxidative stress and regulating apoptosis (cell death).406 Oxidative stress, itself, triggers the activation of FoxO and FoxO, in turn, increases transcription of antioxidant enzymes such as selenoprotein P and manganese-superoxide (SOD1).
This may seem harmless (or even beneficial) on the surface, but consider that selenoprotein P is frequently seen alongside amyloid plaques, so this process cannot be assumed to be “intelligent” enough to halt Alzheimer’s progression. Nature is essentially blind – it does its best with what it has, but in the case of dementia, there are many firemen with a quickly spreading, self-perpetuating blaze.
The truth of the matter is that sustained activation of FoxO plunges the brain into a pro-apoptotic profile that prefers neuronal death over antioxidant rescue.407 To be clear, chronic invocation of gluconeogenesis via FoxO transcription factors leads to insulin resistance, increased oxidative stress, and a shift in FoxO’s transcriptional potential to favor the death of neurons.
In other words, the more you flirt with gluconeogenesis, the greater your potential for brain damage and dementia. Is the need for weight loss or cognitive improvement worth that risk?
FoxO induction of gluconeogenesis, enhanced by octanoic acid, has yet another troubling outcome. If you have any degree of elevated blood sugar and you are also consuming high levels of fat and protein, then you will also be at higher risk for “advanced glycation end-products” or AGEs. These are the proverbial “Tazmanian devils” in the bloodstream that damage everything in their path as they move through the cardiovascular system. In fact, AGEs are likely responsible for the majority of atherosclerosis and heart attacks in the world. It’s not the cholesterol itself, but how that cholesterol is oxidized (or any fat is oxidized, for that matter) and the damage the oxidized lipids can inflict to endothelial tissue. For that matter, AGEs are particularly lethal to neurons!408
Therefore, FoxO signaling is characterized by a vicious circle that begins with chronic gluconeogenesis, proceeds to oxidative stress, and results in neuronal damage. It is self-perpetuating and hard to reverse once the cycle has been established. Fasting with MCTs? Your chances of dementia are not only higher, they are guaranteed. And lest you believe that resistance training, superior insulin sensitivity, and ample glycogen storage will save you, consider the potential variations in the genetics that control glycogen storage, glycogenolysis, insulin secretion, insulin receptor expression, and yes – all the other immunological consequences I have painstakingly outlined above. Are you completely confident that even a tablespoon of these unnatural, artificially extracted triglycerides is in your favor? For that matter, is even coconut oil completely safe?
A Personal MCT Experience
In conclusion, I would like to briefly share my own story about MCTs. Several years ago, I was also enamored by the countless, glowing studies that were being shared by certain biohackers that praise the benefits of dietary consumption of MCT oil. At the time, I was a few years past my 40th birthday and dealing with increasing body fat and a gradually elevating blood glucose. I saw a cyclical ketogenic diet that incorporates MCT oil, mostly caprylic acid, as a solution to these metabolic issues.
To make a long story short, I was not an incredibly healthy child. My father was 50 years old when I was born, and children of older parents are known to have certain immunological challenges as a result of the mutations that occur in sperm over time. This problem, of course, is compounded if the mother is also older, as female eggs also endure such mutagenic changes in DNA. That being said, I was often afflicted by bacterial and viral infections accompanied by very high fevers, sometimes up to 106 or more. I was given the most potent antibiotics available at the time.
Over the years, I gradually came to understand what kind of changes happen in the gut when both pathogenic and commensal microflora are eliminated with broad-spectrum antibiotics. That subject is beyond the scope of this article, but suffice it to say that chronic antibiotic use can and does lead to a further weakened immune system and heightened risk for gut barrier permeability.
Within just a month of using MCT oil, I plunged into perhaps the most frightening health crisis of my life. I was experiencing spontaneous, severe hypoglycemia, elevated ammonia in the brain (verified by testing), neurotransmitter disruptions which bordered on psychosis, and worst of all, hypothyroidism with sporadic dips in energy. Never in my life had I ever been sensitive to cold temperatures, and within just one month, I had to wear a sweater at just 70 degrees Fahrenheit. Fortunately, I can laugh about this now, but at the time, it was a serious, life-threatening problem. And unfortunately, no-one in the community of MCT proponents had anything useful to say about it other than it was possible the oils could be provoking a Herxheimer reaction by killing pathogens in the gut.
Unfortunately, I persisted with the MCT ketogenic diet, despite symptoms – for many reasons, but mostly because I believed it was improving my cognitive ability and giving me a clearer mind. In retrospect, it is quite impressive the lengths we can go to protect our health regimens when dogma regarding their benefits are strong. One practitioner even praised my protocol, assuring me that I would “be guaranteed to avoid Parkinson’s disease later in life”. Nonetheless, though I had lost 30 pounds in a very short period of time and was at a weight I had not “enjoyed” since 20 years of age, I simply didn’t feel well at all. After nearly 2 years of consuming MCTs on and off to varying degrees, I started to notice a slight tremor in my right hand. This marked the beginning of over 5 years of intense research into mitochondria, fatty acids, and neurodegeneration that eventually led to the content I have shared in this article.
Birdseye View: How MCTs Cause Dementia
 Many people have come to me the past several years with one or another problem that can be directly attributed to MCT consumption. Truly, I would not have known to recognize their symptoms had I not experienced many of them first-hand. One thing is quite certain to me now (and hopefully should be abundantly clear to you as well): capric and caprylic acids, in any amount, can produce unpredictable and potentially dangerous effects in the gut, brain, and immune system and do not produce benefits that can justify their risks. To summarize, C8 and C10 triglycerides:
Many people have come to me the past several years with one or another problem that can be directly attributed to MCT consumption. Truly, I would not have known to recognize their symptoms had I not experienced many of them first-hand. One thing is quite certain to me now (and hopefully should be abundantly clear to you as well): capric and caprylic acids, in any amount, can produce unpredictable and potentially dangerous effects in the gut, brain, and immune system and do not produce benefits that can justify their risks. To summarize, C8 and C10 triglycerides:
- Deplete carnitine body-wide and increase risk for fatty-acid induced oxidative stress in the intestinal epithelium
- With carnitine depletion, further have the potential of disrupting the urea cycle and leading to elevations in neurotoxic ammonia (particularly C10)
- Disrupt mitochondrial integrity in the choroid plexus, leading to entry of unwanted molecules, including endotoxins, into cerebrospinal fluid
- Directly increase allergic sensitivity to antigens from food, bacteria, viruses and parasites on all epithelial surfaces, including skin, lung, and gut
- Compromise tight junctions in both the gut and blood-brain barrier, creating wide-radius pores that permit entrance to unwanted molecules (especially toxins)
- With increased consumption, can accumulate in the brain (especially C10), inhibiting energy metabolism in the cerebral cortex, and ultimately disrupting long-term potentiation in an AMPA receptor-dependent manner
- Provoke TSLP-modulated dendritic cells in astrocytes and glia to increase inflammatory activity, oxidative stress, and eventually neuronal damage
- At lower levels, increase calcium uptake in the gut and, in the brain, increase calcium influx at neuronal membranes, elevating synaptic potential and raising the risk for excitotoxicity
- Induce mitochondrial permeability via “transition pores” in a volume dependent manner, leading to mitochondrial swelling and, potentially, apoptosis
- Inhibit reduction of S-nitrosated glutathione, resulting in higher neuroinflammatory nitric oxide signaling in the brain and lower glutamate receptor inhibition (by limited conversion to oxidized glutathione) — higher neuronal nitric oxide synthase expression induces nitrosative stress and further neuronal damage
- Stimulate immune cells and expression of adhesion and granulation markers, inflicting serious effects on leukocyte functions and a lower ability for bacterial killing, regardless of their disruptions to bacterial biofilms, especially in those that are immune-compromised
- Are taken up by colorectal cancer cells to support more efficient metastasis and potentially serve as alternative, non-glucose energy supply for tumors
- At higher levels, burden glucuronidation in the liver, increasing the potential for accumulation of steroid hormones, bilirubin, and retinoic acid – which will also potentiate MCT and MCT metabolite accumulation and further carnitine depletion
- Promote gluconeogenesis in the liver (particularly C8 / octanoic acid), especially in ketotic or fasting states, leading to consistently high blood sugar, shifts in FoxO signaling, and eventually, FoxO mediated apoptotic programs that kill neurons
From mouth, to gut, to brain, medium-chain triglycerides are silently inducing neurodegeneration the world over, and the consumption trend does not seem to show any signs of slowing down. On the contrary, we are seeing an increasing number of products on grocery store shelves and internet marketplaces that contain MCTs, and both the press and social media have been mostly supportive of their use. Unfortunately, the science has been fragmented and lacking in coordination across subject areas, in part due to a lack of funding and low pharmaceutical revenue potential in therapeutic contexts.
Final Perspectives
As should be obvious by now, I do not support the use of MCT oil in any quantity, except for epilepsy therapy – and even in that context, I believe there are better, more sustainable options we can develop. Though many of you that consume MCT oil are completely asymptomatic and can even claim vast improvements in metabolic markers, weight loss, and even increased cognitive agility, I can assure you that these gains are not permanent. Sooner or later, anyone consuming these triglycerides will succumb to one or more of the problems I’ve listed in this article, not the least of which is early onset dementia. Only those that are minding their metabolism in minute detail (and deeply understand their individual genetics) could be considered to have a fighting chance, but the risks are clear and severe. As I will share in future articles, there are far better options for weight loss, cognitive enhancement, and energy balance that carry vastly fewer risks.
If you are over 40 years of age, I strongly urge you to reconsider your position on this matter and do what you can to educate yourself and come to your own conclusions about it. This will allow you to make mindful and appropriate decisions that will protect you and your brain for years to come. My only hope is that the community at large does not wait another 20-30 years to raise the red flag on these risks. It will take about that long for epidemiological data to reflect an issue, but by then, the experts will be pointing the finger at other causes. Few will suspect MCT as the smoking gun, given the complexity and confounding factors inherit in epidemiological data.
While we can expect significant advances in the field of neurology, especially with the aid of artificial intelligence and data science, this should not embolden us to take a risk with our bodies based nothing more on subjective feelings of well-being and a handful of cherry-picked studies proving benefits. We must consider everything from all possible angles, without personal bias or emotional / political influences. Hopefully, I have at least marginally accomplished that task in this article.
Do you have an MCT story?
If you have personally been affected by one or more the symptoms that I have described here, I encourage you to reach out to me to talk about your options. The more advanced symptoms are, the more aggressive and thorough the interventions need to set things right again. Don’t wait – take action now! At the very least, please think twice the next time you hold a bottle of “brain fuel” in your hand, preparing to pour it into your coffee or onto your food.
Join our program today!
Transcend Genomics offers an unparalleled opportunity for self-exploration, growth, and radical body-mind optimization. The journey begins with an analysis of over 20,000 genes and 200 metabolic pathways to discover the areas of your genome that could have the greatest impact on your health, cognition, and longevity.