

In late 2019, a mysterious outbreak of pneumonia occurred in Wuhan, Hubei Province, China, eventually prompting an announcement, on New Year’s Eve, by the Chinese Center for Disease Control and Prevention. A growing number of patients were presenting to hospitals with severe symptoms, most of which had visited the Huanan Seafood Wholesale Market.12
Expedient efforts were made to contain transmission of what became known as SARS-Coronavirus-2, but infection rates far outstripped the best strategies, and new cases started cropping up in Thailand, Japan, and South Korea.34
It wasn’t until January 7, 2020 that the Chinese CDC had been able to classify this new coronavirus from samples extracted from the lower respiratory tract of patients suffering from pneumonia. The full genomic sequence for the virus was revealed just a few days later on January 11. The World Health Organization (WHO) later named the infection caused by SARS-CoV-2 Coronavirus Disease 2019 (COVID-19).
This was not our first encounter with the coronavirus family. Over the past two decades, there were two other major outbreaks including SARS-CoV-1 in 2002-2003 and MERS-CoV (Middle-East Respiratory Syndrome) between 2012 and 2019. Deaths from SARS and MERS were in the order of 774 and 861, respectively, whereas the new, more severe SARS-CoV-2 has, at the time of this article’s publication, already taken nearly 300,000 lives. We are currently amidst a full-blown pandemic, and there is no question that we are dealing with a pathogen that is far more contagious and life-threatening than any others in its family that has come before it.5
Unfortunately, as a result of the global health crisis and multiple unknowns surrounding the pathology and mutation rates of the virus, a growing sense of unrest has gripped the world. This has led to an overabundance of conspiracy theories, incomplete conclusions about therapeutic options, and, worst of all, a surge in unfiltered, social media-sourced advice regarding how to protect our health.
Therefore, the purpose of this article will be to lay out, as clearly and concisely as possible, all relevant, scientifically validated points of view established to date regarding SARS-CoV-2 origin, pathology, and risks as well as potential therapeutic strategies.
This article has been structured as follows:
-
We will begin with a 10,000-foot view of the current pandemic followed by my interpretation, based on currently known facts, of the stages of disease and its progression. We will discuss why some people experience acute respiratory distress (ARDS) of unparalleled severity while others are relatively symptom-free.
-
Building upon this disease progression framework, I will then elaborate first on current medical interventions and their benefits and risks followed by a discussion of novel therapies now being proposed on the cutting edge of modern medical technology.
-
We will then turn our attention to natural approaches now being freely recommended, mostly by functional medical practitioners but also by what is commonly known as the “alternative medical movement”. Myths and misconceptions regarding melatonin, ascorbic acid, vitamin D, and other such “silver bullets” will be analyzed, dissected, and presented to better help you understand the risks of embracing such approaches while also considering their potential as adjuncts in more scientifically grounded therapies.
-
Last but not least, I will be sharing for the first time the results of our own predictive model for both pharmaceutical and natural compounds using our patent-pending BiopathFx methodology. Using inference networks, we have derived strategies that combine the best of both worlds and propose their use in a multi-pronged approach that considers individual genetics at every potential stage of COVID-19 progression.
This article could have been written much earlier in the course of the COVID-19 pandemic. Much of it had been completed late January, but as research progressively surfaced around the world, it became clear to me that many more facts were needed to come to useful conclusions about this disease’s pathology and needed strategies for resolution. I decided that it would be best to take more time to observe the opinions of medical experts, scientists, and clinicians around the world in order to more deeply understand the challenges that COVID-19 presents.
This article is the result of months of in-depth research covering hundreds of biological pathways and over 1,500 scientific papers spanning the last two decades and beyond. As such, it is my desire that this will be the most comprehensive and thorough treatise on this pandemic and that this information will be used to develop effective, customized strategies rather than scientifically unviable, one-size-fits-all approaches.
Birdseye View: The Epidemic
 Origins and Mutation
Origins and Mutation
One conspiracy theory that nearly everyone is familiar with involves the claim that SARS-CoV-2 was engineered in a lab with the intention of purposely making people sick and/or influencing the global economy. Apart from the fact that this idea is severely unpractical, it has already been thoroughly debunked by a group of scientists from Scripps Research Institute and Tulane University.
In March, 2020, along with their colleagues, Kristian Andersen and Robert Garry provided evidence that SARS-CoV-2 arose naturally. Using advanced bioinformatics, they compared the new virus with genomic data from pre-existing coronavirus strains.6
What they found were progressive, evolution-driven changes to the spike proteins that give the coronaviruses their distinctive “crown”. Though all of the viruses in this family use the spike proteins to infect host cells, the proteins’ genetic code has been modified over time to allow more effective cell entry and infiltration.
SARS-CoV-2 spike proteins, in particular, share similarities with those of the coronavirus that caused the SARS epidemic of 2002-2003. Both viruses bind to the angiotensin-converting enzyme (ACE2) receptor on cell surfaces to facilitate rapid entry. CoV-2 is different in that it has modified its spike proteins to utilize an alternative binding site on the ACE2 receptor, allowing it a better stronghold.
Any skilled bioengineer would immediately recognize that this spike protein adaptation is a result of natural selection. If the intention were to create a maximally lethal laboratory-derived virus, there are other receptors and binding patterns that would be far more effective. The use of the same ACE2 receptor targeted by SARS-CoV in 2002-2003 indicates not only that this is a relative of that branch of coronavirus but perhaps even a direct adaptation of the same strain.
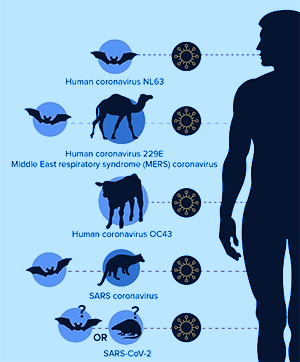
Further, were the manufacture of a virally-based bio-weapon to be the ultimate goal, the scientists would have utilized backbone structures of other coronaviruses known to be far more dangerous to humans. On the contrary, the Scripps / Tufts analysis revealed the SARS-CoV-2 backbone to coincide with that of another strain found primarily in bats. The ACE2-binding region, specifically, resembles yet another strain found in pangolins. And it has been conjectured that the jump from bats to humans most likely occurred in late November or early December 2019.7
In other words, the virus that provoked COVID-19 arose naturally in the animal kingdom and has no precedent in human models of infection. Were there to be a synthetic, lab-derived origin for this virus, far better models known to be dangerous to humans would have been chosen. In fact, receptor-binding type mutations transitioning from the animals to humans is not new to the coronavirus family. This pattern was also seen in SARS and MERS, which arose from civets and camels, respectively.
Another theory exists that SARS-CoV-2 transitioned to humans long ago and, over the course of time, mutated within our species in order to adapt to our specific cellular receptor types. We will perhaps never know, but one thing is certain: the human race is in this together, and a malicious group of criminals did not pre-meditatively provoke the COVID-19 crisis in a laboratory.
Incubation, Contagion, and Transmission
Incidence of COVID-19 continues to increase worldwide, despite extensive containment and quarantine strategies. Epidemiological studies have demonstrated 1.4 to 3.9 new cases stemming from each documented infection, though these numbers relate to communities with either zero immunity or adherence to proper prevention. The epidemic is expected to continue until we can get this number down below 1.8
Early reports estimated the incubation period for this virus to be ~5 days. However, recent data shows that symptoms may be delayed as long as 12-14 days, considerably widening the asymptomatic contagion window.910 Contagion potential peaks roughly 3 days after the onset of symptoms, depending on viral load.
Though viral loads are expected to decline with time, immunocompromised individuals may be contagious for longer periods of time due to lower capability for viral control. It is therefore extremely important to consider the immune status of an infected person when determining the period of convalescence.11
There has been a lot of debate over the specific mechanism of transmission. It is generally accepted, however, that human-to-human transmission occurs primarily via droplets. One study performed in Singapore discovered that an uncovered cough can lead to droplets that travel as far as 4.5 meters (15 feet). This is been the subject of rigorous debate as well, as yet another study argued that the warm, moist out-breath surrounding the droplets after an uncovered cough or sneeze could potentially travel more than 8 meters. Considering that’s the distance of almost five 6-foot tall individuals lined up head to toe, the 6ft social distancing guidance comes squarely into question.121314
Transmission and infection occurs when the droplets land in the mouths or noses of those close by and/or inhaled into the lungs. It is also well known at this point that apart from coughing and sneezing, people can also become infected by first touching contaminated surfaces and then touching their eyes, nose, or mouth. Unfortunately, droplet-type transmission is not limited to those displaying obvious symptoms. Respiratory droplets are also produced while breathing and talking, in general.
As a matter of fact, the New England Journal of Medicine reported earlier this year that SARS-CoV-2 may be capable of floating in aerosol droplets of less than 5 microns wide as long as 3 hours, while remaining infectious. They cited a University of Nebraska paper showing widespread evidence of viral RNA in isolation rooms of COVID-19 patients. This RNA was not only on hard-to-reach surfaces, but in air sampling devices as far as 2 meters from the patient. If you read the fine print of that last study, however, the collected particles were not found to be contagious.1516
There has been some talk about the potential for fecal-to-oral transmissions. Existing scientific literature on the subject has shown SARS and MERS to be viable in oral-fecal transmissions, but similar evidence has not been established for SARS-CoV-2.1718 Nonetheless, SARS-CoV-2 RNA has been detected in stool, whole blood, and urine. Keep in mind this does not mean that exposure to such mediums equates to infection.1920 The good news is that SARS-CoV-2 appears to be relatively fragile, especially with exposures to temps above 27C / 80.6F.
Unfortunately, it has variable survival rates on different surfaces: one day on cardboard, four hours on 99% copper, and three days on plastic / stainless steel. Caution is advised when touching such surfaces if they have come in contact with someone whose activity cannot be accounted for.2122
 Demographics
Demographics
Many of the genes that confer immunological resilience are located on the X chromosome, giving women a clear advantage in the risk for CoV-19 susceptibility. Not surprisingly, more than 50% of documented cases have been male.23
Only a few cases have been attributed to children under the age of 15. Many theories have been floating around, explaining this phenomenon, but the average immunologist will tell you this is connected with the greater size and efficiency of the thymus in early age. Younger children are further afforded protection by maternal immunity.24
According to the Chinese National Reporting System, as of February 20, 2020, the median age of the confirmed cases was 51 years, of which 77.8% were 30-69 years.25 The risk factors for severe pneumonia and acute respiratory distress increase with both age and pre-existing conditions such as diabetes, hypertension, cardiovascular disease, or other pulmonary disorder like asthma or COPD.
Diagnosis
Currently, diagnosis of SARS-CoV-2 is being performed via nucleic acid amplification tests such as reverse real-time PCR (rRT-PCR), taken from a nasopharyngeal swab.26
Not all tests are created equal, even among real-time PCR assays, and those that focus on the RdRp/Hel gene of the virus have shown higher sensitivity than those that use RdRp-P2. Therefore, the statistics coming in for confirmed cases is not likely to be 100% reliable. Nonetheless, rRT-PCR is considered to be at least 90% accurate across the board, with some assays reaching a full 100%.27
 Prevention (and the truth about masks)
Prevention (and the truth about masks)
Much has already been said on the topic of prevention, including the advice to thoroughly wash hands and avoid touching the face. What may not be explicitly obvious for many people, however, is that even masks do not completely mitigate the risk for transmission and infection.28
Ultimately, only quarantine and strict containment strategies can fully isolate infected individuals and keep them from spreading the virus. Given the extreme disruptions this can (and has) caused in our global economy, it would make sense to delve more deeply into the wide variability in protection that masks can afford the public at large.
Masks may become the single most important factor in the containment of this pandemic, given that most world leaders have expressed desires to lift restrictions and re-open businesses. As I’ve mentioned above, until we have achieved a per case infection rate of less than one, lifting restrictions can only increase the potential for a second wave of infection, despite rising temperatures. And while there will most certainly be progressively more effective therapies deployed in the future, we cannot yet predict how well they will achieve complete containment of this problem.
The primary issue with using masks for COVID-19 containment is the SARS-CoV-2 particle size, which ranges from .07 to .09 microns. Event the robust N95 respirators only block 95% of particles, with the smallest size being .3 microns. N99 masks are not much better, with an identical particle size limit and 99% efficacy. The largest SARS-CoV-2 particle is already 3 times smaller than the lower limit for both of these masks.2930
Unfortunately, surgical masks are even less effective, given their low efficacy against particles between .04 and .2 microns.3132 One observational study in China demonstrated infections in 10 out of 213 medical staff without masks, whereas another group showed 0 out of 278 staff using the N95. Therefore, even with its inefficient filtration for SARS-CoV-2 sized particles, these makes are nonetheless efficient in clinical environments.33 For this reason, they are the mask of choice for healthcare workers, especially considering that cloth masks have been shown to be 13 times less likely to protect against flu-like illness.34
Many people around the world have been using surgical masks when going out in public, and though they have shown the ability to inhibit cold and flu transmission for sick people, they haven’t truly proven effective for the healthy population.35 One study showed 97% penetration for cloth masks as opposed to the 44% with surgical masks, so while the surgical mask may be the better option, one shouldn’t be emboldened to endure prolonged exposures to infected individuals or spend time in larger groups of people.3637
Here, it’s important to be reminded that the primary transmission route for SARS-CoV-2 is prolonged close contact with infected individuals. In such contexts, N95 masks or better would be considered mandatory. That being said, there has been a problem with people purchasing such masks for personal use, creating shortages in clinics and hospitals where they are needed more. This is one of the problems with publicly talking about and comparing filtration properties of masks, in general. The reader may automatically presume that cloth or surgical masks are completely useless. I want to be very clear that they are marginally useful in urban contexts with zero prolonged exposures to infected people, and wearing one in a crowded environment would be expected to have some mitigating effect on transmission.38 Nonetheless, a randomized trial that had four COVID-19 patients cough into a petri dish while wearing various types of masks was extremely disappointing. Neither the surgical nor the cloth masks did very well at all filtering SARS-CoV-2.39
Therefore, my stance on masks is that they are effective at slowing the spread of CoV-2 when worn by a sick individual, but will not necessarily afford direct protection for a healthy one. In a sense, there is a certain amount of protection by using a mask, given the lower likelihood that one would be directly touching the face, but just be aware that CoV-2 particles can and do make their way through both cotton / surgical masks as well as N95 / 99 respirators. Again, the only true prevention is isolation until an effective therapy has been developed and implemented. I’ll be talking in more detail about such therapies further below.
 Variability of Symptoms and Severity
Variability of Symptoms and Severity
A wide spectrum of symptoms have been described for COVID-19, ranging from mild cough and fever to full-blown pneumonia, acute respiratory distress, and even cardiac arrest. Multiple factors play into the risk for specific symptoms, depending on which stage of the pathology a person is in.
In up to 10% of cases documented thus far, fever and respiratory symptoms are preceded by diarrhea and nausea. As I will describe below, this is in part due to the presence of ACE2 receptors in glandular cells of gastric, duodenal and rectal epithelium as well as endothelial cells and enterocytes of the small intestine.404142 Roughly 101 out of 10,000 cases of COVID-19 will develop symptoms within 14 days of observation or quarantine.43 The most common symptoms are fever, cough, and shortness of breath, seen in over 30% of all cases.44
Generally, patients that develop acute respiratory distress syndrome often experience rapid decline, followed by multiple organ failure. This process can be exacerbated by using the wrong medications at the wrong time. In 25-30% of all patients that have been admitted to the ICU, there is a high incidence of thrombosis and thromboembolism, due to blood vessel dysfunction and clotting irregularities. Such problems are the result of COVID-19 provoked vascular inflammation and subsequent derangement of hemodynamics (i.e. blood cell behavior).
There is wild variability in the pathology for COVID-19 from person to person, but the common thread is massive virally mediated inflammation in the epithelial cells of the lung followed by vascular permeability, fluid accumulation in alveoli, surfactant dilution and alveolar collapse, and compromised gas exchange. At the time of hospitalization, neutrophils are already being recruited to infection sites, increasing oxidative damage, and pneumocytes are gradually destroyed, resulting in collapse of alveoli and eventually, acute respiratory distress syndrome (ARDS). If the inflammation becomes severe enough, protein-rich fluid may enter circulation and migrate to other parts of the body, inducing sepsis. In the worst-case scenario, this can lead to septic shock and death.
While there is also the potential for red blood cell rupture (hemolysis) and a disruption in oxygen-carrying potential for hemoglobin, there has been no concrete clinical documentation proving this is a significant part of the ARDS pathology. On the contrary, biomarkers of excessive cell-free hemoglobin as observed, for example, in HBOC transfusions have not been demonstrated in COVID-19 patients, and a deeper examination of the pathology and its lab markers would confirm that hemolysis does not play a substantial role in disease progression. Nonetheless, I will be describing its potential, as a great deal of pseudoscience has recently developed around the importance of cell-free hemoglobin in COVID-19 ARDS. That being said, certain individuals may be more vulnerable to hemolysis and additional oxidative stress from cell-free hemoglobin metabolic products, so we will include this pathology in our discussion of disease stages
 Death Rates and Autopsy Findings
Death Rates and Autopsy Findings
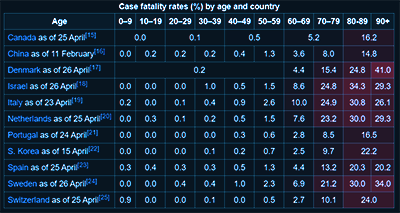
In general, intensive care is required for 20 to 30% of all patients that present with COVID-19 symptoms, and there has been a 10% fatality rate. In China, the death rate has been 2.8% for men and 1.7% for women. For studies conducted in Italy, mortality has been higher in men, especially those in their 50s. In fact, the gap between men and women has not been shown to close until the age of 90.45
So far, there have been no recorded COVID-19 fatalities in patients under the age of nine. Fatality increases linearly with age, with the highest rate being seen in individuals over the age of 80. Experts disagree as to why young children are spared from COVID-19, but as I have mentioned above, immunologists tend to believe that greater thymic size and activity are one of the primary reasons.
My personal opinion is that COVID-19 immunity is not simply about the thymus and spleen but, rather, rooted in progenitors for all blood cell types. The reason children (and younger humans in general) are afforded greater protections from COVID-19 complications is because of more dynamic and resilient blood cell differentiation in youth. Some unofficial information sources have proposed higher melatonin levels in youth (and pregnancy) as protective, but such concepts, as I will describe in more detail below, are not founded in a deeper understanding of immunology — or circadian hormone regulation, for that matter. The hematopoietic cell linage, in fact, is the primary driver of immune status and gradually goes offline with age, starting as early as adolescence in some people. Derived from bone marrow, this lineage gives rise to myeloid and lymphoid progenitors, both of which are unimaginably stressed during COVID-19 disease progression. In young people, there is balanced linage output (i.e. even distribution of erythrocytes and lymphocytes), higher regeneration potential/self-renewal, and polyclonal hematopoiesis.
COVID-19 puts tremendous stress on both myeloid and lymphoid blood cell lineages, generating massive oxidative stress, mitochondrial dysfunction, and systemic hypoxia (in the later stages). For older individuals, there are pre-existing myeloid imbalances resulting in skewed erythrocyte, mast cell, and myeloblast (i.e. leukocyte) populations. This is exacerbated by lymphoid deficiency resulting in lower natural killer, T, and B cell numbers as well.
 Potential Therapies
Potential Therapies
COVID-19 is a complicated disease with new challenges we have not seen before. Hundreds of potential therapies have been considered, and Korean and Chinese health authorities have already recommended the use of the anti-malarial drug chloroquine up to 1 gram per day. Unfortunately, this drug is far from being a silver bullet and carries some very significant health risks, depending on individual genetics and other pre-existing conditions. One Brazilian study, for example, was halted upon the discovery that chloroquine can be lethal at high doses.46
Currently, there are more than 300 active clinical trials underway, reviewing potential candidates such as vasodilators, immune therapies, corticosteroids, ACE inhibitors, and more.47 Vaccines have been proposed, and on March 16, 2020, the first clinical trial of a vaccine began with four volunteers in Seattle.48
Later in this article, there will be a more exhaustive review of both pharmaceutical and natural compounds along with their pros and cons as therapeutics. First, let’s dive more deeply into the COVID-19 pathology, to better understand what makes it so unique.
COVID-19 Pathology: A Detailed Look
 In the less severe cases of COVID-19, the disease behaves similarly to an upper respiratory tract infection not unlike the common cold. In the presence of more rapid viral replication, however, the disease begins to take on the quality of severe acute respiratory syndrome marked by widespread infection of the lower airways. This leads to massive pro-inflammatory cytokine release and potentially fatal pneumonia, particularly for those suffering from other pre-existing conditions such as hypertension, diabetes, or asthma / COPD.49
In the less severe cases of COVID-19, the disease behaves similarly to an upper respiratory tract infection not unlike the common cold. In the presence of more rapid viral replication, however, the disease begins to take on the quality of severe acute respiratory syndrome marked by widespread infection of the lower airways. This leads to massive pro-inflammatory cytokine release and potentially fatal pneumonia, particularly for those suffering from other pre-existing conditions such as hypertension, diabetes, or asthma / COPD.49
Medical workers in the field have described tragic outcomes connected with the use of ventilators which only exacerbate lung damage due to increased internal pulmonary pressure. The longer a person stays connected to such machines, the greater the pressure-induced destruction of alveoli. This results in “lung remodeling” — a progressive buildup of scar tissue, further narrowing airways. Nurses and doctors have described perfectly healthy 40-year-olds holding steady on low amounts of oxygen only to be on the verge of respiratory arrest in just a matter of hours.
Typically, ARDS gradually progresses in parallel with lung inflammation. SARS-CoV-2, however, infiltrates far more rapidly than its predecessors, exponentially speeding up the pro-inflammatory cascade. By injuring cells in the walls of the alveoli, fluid and even blood can leak in. In the process of passing through inflamed tissue, red blood cells may even rupture, releasing free hemoglobin which can quickly oxidize and create even greater levels of inflammation. As fluid accumulates in the lungs, there is the appearance of the “ground-glass opacity” described on the X-rays.
Medical professionals in ICU’s have described having to engage the ventilators at maximum capacity (i.e. 90% oxygen and level 16 positive end-expiratory pressure / PEEP) in order to keep lungs inflated. This level of pressure is rarely needed in other contexts, and many doctors and nurses have never had to use it before.50
In order to understand how SARS-CoV-2 can induce such rapid damage and lead to ARDS in vulnerable individuals, it would be useful to briefly review basic lung anatomy and the immunological response in epithelial cells.51
Anatomy of the Lungs: A Brief Review
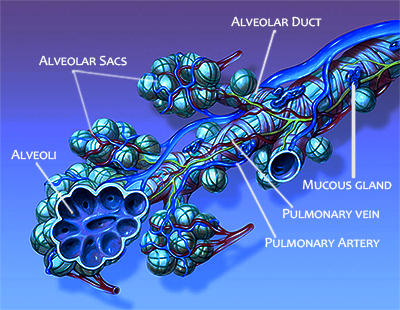 When breathing in air saturated with viral RNA-containing droplets, some of those droplets adhere to the top of the respiratory tract at the trachea. Those that continue on will make their way through the bronchi and bronchioles, eventually ending up in the alveolar ducts, terminating in alveoli containing sacs. It is within the sacs that bidirectional exchanges of oxygen and carbon dioxide take place.
When breathing in air saturated with viral RNA-containing droplets, some of those droplets adhere to the top of the respiratory tract at the trachea. Those that continue on will make their way through the bronchi and bronchioles, eventually ending up in the alveolar ducts, terminating in alveoli containing sacs. It is within the sacs that bidirectional exchanges of oxygen and carbon dioxide take place.
The entire respiratory tract, from the trachea to the bronchioles, is lined with respiratory epithelium interspersed with mucin-producing goblet cells, hair-like “cilia”, and special stem-cells (i.e. basal cells) capable of regenerating the epithelium during cell turnover or after sustaining damage. In the terminal bronchioles, macrophages may also be found. These macrophages are the first line of defense against pathogens that have made their way into the terminal bronchioles and autopsies often reveal that they contain black specks of concentrated carbon accumulated from, for example, automobile exhaust and other urban pollutants.
All of these epithelial cells, including submucosal glands throughout the entire respiratory tract, secrete airway surface liquid (ASL). These secretions are very tightly regulated, and any aberrations in function can hinder the ability of the mucociliary “machinery” to clear particulates and other invaders for expectoration in mucus.
There are two circulatory systems that carry blood to and from the lungs which are important to visualize as part of our deeper understanding of ARDS in COVID-19. The first system is pulmonary circulation which carries deoxygenated blood away from the right ventricle of the heart to the lung alveoli, where it releases carbon dioxide in exchange for oxygen. It then returns the oxygenated blood to the left atrium and ventricle of the heart where it may be pumped out to the rest of the body.
The second system is bronchial circulation, which supplies fully oxygenated arterial blood to lung tissues such as bronchi and pleura in order to meet their nutritional requirements.
 Alveoli: Two Types
Alveoli: Two Types
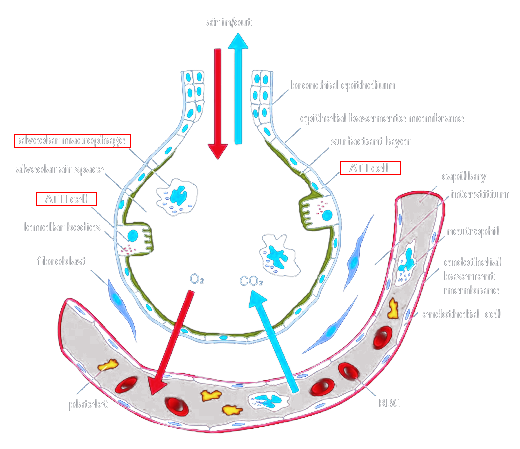
Alveolar cells at the end of bronchioles are the most important component in the process of gas exchange in the lungs. There are three types of these cells. The first two types are known as type I and type II pneumocytes.
Type I pneumocytes are squamous (i.e. “flat”) and comprise the alveolar wall structure and septa. These thin walls easily permit the exchange of gas due to their increased surface area. Unfortunately, they are also more vulnerable to damage. To make matters worse, they are not able to divide and rely completely on differentiation from Type II pneumocytes.
Type II pneumocytes, themselves, are quite a bit larger and outnumber all other cells in the alveoli. They coat the alveoli, producing fluid and phospholipid-containing surfactant. The fluid facilitates the transfer of gases between blood and alveolar air, and the surfactant reduces surface tension, thereby making it easier for the lungs to reinflate after exhalation. Even though Type II cells are slightly more robust than their Type I children, they can nonetheless be damaged, causing the lungs to be even more vulnerable to insult.
The third type of pneumocyte, and perhaps the most important in the ARDS pathology, are the alveolar macrophages, which reside on the internal lumenal surfaces and ducts of the alveoli. Similar to the macrophages found in the terminal bronchioles, these alveolar immune cells remove particle deposits in alveoli such as dust, bacteria, carbon particles, and blood cells that have been forced out of blood vessels due to vascular damage or increased permeability.
 Lung microbiome
Lung microbiome
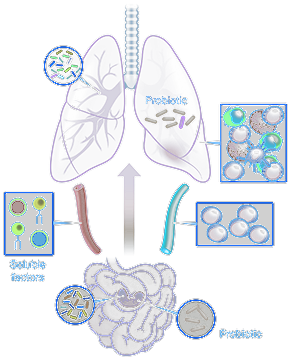
Apart from pneumocytes and macrophages, there are a large number of other microorganisms that exist throughout all lung structures that comprise what has become known as the “lung microbiome”. Similar to the microbiome in the gut, microbiota in the lung have multiple interactions with airway epithelial cells. In fact, the level of microbial diversity in the lung is directly correlated with the degree of respiratory health.
As such, any infection that targets the lungs, including the common rhinovirus, can significantly change the microbial populations. Therefore, the status of the lung microbiome has far-reaching implications for immunological response to respiratory infections, especially of the virulent type such as SARS-CoV-2.52
Immune Dysregulation in Epithelial cells
Understanding microbe-epithelial cell interactions in the lung are vital to a full comprehension of how immune dysregulation takes place in the context of infection. Airway epithelial cells (AECs) are literally the first line of defense in the lungs against inhaled gases, particles, and microbial pathogens. It wasn’t long ago that medical science viewed AECs as nothing more than a structural barrier that clears inhaled substances via mucociliary action. We knew that this barrier regulates water and ion transport, but it wasn’t until recently that AECs were discovered to play vitally important roles in immunological defense, inflammation, and even tissue remodeling processes such as fibrosis.
Among the functions that AECs perform include the production of antimicrobial peptides and proteins, reactive oxygen and nitrogen species (ROS/RNS), and a broad range of cytokines, chemokines, and growth factors.5354 Normally, the lumen of the airways is protected from inhaled pollutants, microbes and allergens by a barrier comprised of tight and adherens junctions — identical in function to that found in the GI tract. However, if the AECs are continuously exposed to bacteria, viruses, and other pathogens, they can disturb microbial balance in the lung microbiome.
In recent times, real-time PCR, pyrosequencing, and other methods have shown a disturbed lung microbiome in individuals with asthma and COPD.5556 This confirms observations in other studies showing a link between lung microbiota disruptions and shifts in immunological tolerance to allergens.57
Viral detection mechanisms
AECs are capable of sensing the presence of bacteria and viruses by recognizing molecular structures unique to their envelopes/membranes. These pathogen-associated molecular patterns (PAMPs) are identified by fine-tuned transmembrane proteins on AECs, also known as pattern recognition receptors (PRRs). PRRs recognize PAMPs on microbes, bind to them, and then trigger intra-AEC cellular signals that provoke an immune response.58 Four families of PRRs have been identified. They include Toll-like receptors (TLRs) and C-type lectin receptors (CLRS) in membranes and retinoic acid-inducible gene (RIG)-I-like (RLRs) and the NOD-like receptors (NLRs) in cytoplasm.59
The TLRs, in particular, though found docked in AEC membranes, are far more abundant on the alveolar macrophages.6061 Single-strand RNA viruses such as SARS-CoV-2 are recognized by TLR7 and 8, so depending on an individual’s genetics for TLR7 and 8 expression, there may be a wide variety of immunological reactions (both under- and hyper-responsive).6263
Antiviral defense and inflammation
Speaking of genetics, there is a large host of genes responsible for AEC immunological expression that can have profound impacts on how well an individual is protected against SARS-CoV-2 viral infection. The first line of defense is the discontinuous mucus layer, separated from the epithelial surface by a periciliary or “brush” border. In this layer, cell-tethered mucins are attached to the cilia and form a protected area impenetrable by molecules from above, even mucus.64
This mucus / brush-border complex comprises the airway surface liquid (ASL), and the volume and composition of its secretions are modulated by specialized channels that transport ions and water such as CFTR (cystic fibrosis transmembrane conductance regulator) and ENaC (epithelial sodium channel).65 Should bacteria or viruses manage to make their way past the mucus/brush border, the AECs are capable of further secreting antimicrobial peptides that not only neutralize bacteria and viruses but also modulate inflammation, repair, and regeneration. One such family of peptides, the β-defensins, are induced upon TLR recognition of microbial PAMPs. These “defensins” may also be spontaneously released simply in the presence of pro-inflammatory cytokines.
β-defensins neutralize pathogens in two ways. First, being cationic, they are capable of displacing calcium and magnesium ions in microbial membranes. Due to their greater size, these defensins effectively destabilize the pathogen’s membrane. This changes the membrane’s electrical potential, permitting other defensins to pass through, aggregate, and create pores. These pores in turn depolarize the membrane, breaking open the cell.666768
The second way defensins protect lung infections is by enhancing the adaptive immune system via chemotaxis of monocytes, T-lymphocytes, dendritic cells and mast cells. Defensins have also shown the capacity to improve alveolar macrophage phagocytosis.697071727374
Of great interest to the discussion of COVID-19 is the complete absence of defensin genes in some individuals. Further, the defensin genes show an extremely high rate of polymorphism, specifically in gene copy number, which has been correlated in at least one study to Crohn’s disease in humans.7576 Paradoxically, the longer inflammation is present in the lung, the less likely defensin activity will be present. At least one study focusing on smokers has proven that chronic inflammation, in fact, may very well suppress the expression of antimicrobial peptides.77
Defensins are not the only peptides secreted by AECs. Cathelicidins, such as hCAP-18 and LL-37, have very potent antiviral activity. In the lung, LL-37 is not only produced by AECs but also by neutrophils and macrophages.78 Cathelicidins will be seen playing a more prominent role in later stage acute respiratory distress, given their function in the repair of AECs and promotion of angiogenesis during hypoxia. LL-37, in particular, is acutely produced by neutrophils, but vitamin D is also capable of provoking macrophages to produce it.79
In COVID-19, the higher the viral load, the greater potential for infiltration through the mucus / brush-border complex and induction of chronic inflammatory immune responses. One such response involves AEC-mediated production of highly anti-viral reactive nitrogen species (RNS) by up-regulating nitric oxide synthase (NOS) enzymes. In the airways, nitric oxide production is primarily derived from inducible NOS2 (iNOS).80
In addition to RNS, AECs also produce substantial amounts of reactive oxygen species (ROS) via NADPH oxidases and dual oxidases, collectively known as the NOX/DUOX family.8182 NOX enzymes in lung epithelia are considered to be one of the catalysts for superoxide production, which is potently antimicrobial. As levels rise, superoxide dismutase (SOD) neutralizes excess superoxide by breaking it into oxygen and hydrogen peroxide. Lactoperoxidase can then catalyze the remaining hydrogen peroxide with thiocyanate to form OSCN- (hypothiocyanite), another potent antimicrobial. Should there be any change in genetic expression for NOX, SOD, or lactoperoxidase, there could easily be a build-up of one or more of the intermediates in those reactions leading, on the one hand, to higher antimicrobial potential but, unfortunately, on the other hand, to AEC damage, inflammation, and chronic hypoxia.83
It should come as no surprise that levels of exhaled hydrogen peroxide are correlated with asthma severity, due in part to the elevated ROS production and SOD-mediated activity in hyper-immune asthmatic airways. That being said, it should also evoke great reservations for therapists in the field who are now suggesting nebulized hydrogen peroxide for the treatment of COVID-19. Such a treatment could pour oil on the fire in vulnerable individuals, leading to increased potential for hydroxyl radical formation, especially in the presence of iron ions. Clearly, an individual’s pre-existing condition and genetics for NOX, SOD, and LPO are paramount to understanding where and when to apply specific interventions. We will discuss this concept in much greater depth in the following discussion on disease stages and severity.8485
The next line of defense for AECs against viral infection is via the production of interferons, so-named for their ability to “interfere” with viral replication. COVID-19 has been shown to acutely up-regulate interferon expression, thereby increasing levels of natural killer cells and macrophages.86 In this way, interferons will increase overall inflammation levels and in some cases even suppress the immune system. Nonetheless, they have also been shown to confer protection against autoimmune-type reactions.8788
Though prolonged exposure to interferons can induce apoptosis, suppress cell proliferation, and incur tissue damage, it is generally thought that those individuals that proceed to severe complications from COVID-19 may have inadequate interferon response. For this reason, interferon therapies have been considered in certain contexts, such as COPD, asthma, and cystic fibrosis which have proven to have blunted interferon-mediated host defenses.89 Should viruses make their way into the lung epithelial cell interior, the autophagy program is invoked to help clear toxic components and pathogens from the cytosol by delivering them to lysosomes for degradation. Unfortunately, excessive and uncontrolled autophagy can contribute to further respiratory distress, as has been seen in COPD.90
How asthma increases risk
One of the reasons that pre-existing asthma is such a risk factor for SARS-CoV-2 infection is because of the way that disease alters the airway epithelium by increasing fragility, lowering barrier function, disrupting antioxidant defense, up-regulating basal cell production of goblets, and impairing innate antiviral responses, particularly interferon-mediated ones.91 As a result, there is lower mucociliary clearance of viruses due to excessive mucus production in parallel with compromised antiviral immunity. Further, allergic, Th2-driven cytokine production will all but shut down secretion of antimicrobial peptides such as beta-defensin and LL-37.9293
This leads to higher viral proliferation in lungs, exponentially increased inflammation and, as suggested by one study, a “memory pool” of basal progenitor cells in the epithelium that can, for example, produce excessive amounts of IL33 — an interleukin known to induce helper T cells, mast cells, eosinophils, and basophils to produce type 2 cytokines.94
Stages of Severity
 The Truth about COVID-19 Genetics
The Truth about COVID-19 Genetics
Taking all of these factors into consideration, it should be quite obvious why certain individuals are more prone to serious complications from COVID-19. Immunological genetics and pre-existing conditions play crucial roles in determining not only how severe symptoms could be but also which therapeutics would be most beneficial. In my opinion, individual variability has not been extensively considered so far in the approaches that have been proposed, prior to implementing them for COVID-19 patients. The medical community has been primarily in defensive, reactionary mode, often scrambling for resources to meet demands.
The truth of the matter is that immune-genetics are a crucial talking point in the COVID-19 vaccine debate. Human genomes are highly polymorphic, especially with regards to the chromosomes that carry immune-related genes. As I’ve already mentioned, a great number of those genes are located on the X chromosome, affording women an advantage while also explaining, in part, why men are statistically more likely to suffer complications from this disease.95 Sexual differences aside, genetic variability in immune response has clear correlations with clinical symptoms, disease progression, and overall response to therapies, including vaccines. For this reason, the New York State Department of Health has partnered with it’s Genome Center and Rockefeller University to better understand COVID-19 from a genetic perspective — something we’ve been occupied with extensively here at Transcend Genomics since the onset of the pandemic.96
Categorizing individuals not only by their symptoms but also by specific inflammatory markers combined with rigorous analysis of all relevant immunological genetics — could be the only viable way forward for humanity at large as viruses similar to SARS-CoV-2 appear, mutate, and gain advantages over time. This pandemic has proven to us, beyond a shadow of a doubt, that we are not poised for an effective and adequate response to epidemiological “curve balls”. Make no mistake about it, this is only the beginning for our species, as unnatural pressures in our environment, both natural and self-created, are inevitably changing our immunological resilience and capability for adaptation.
Biomarkers for Mortality
To date, COVID-19 cases have been roughly divided into three groups: ordinary, severe, and critical. The single factor that delineates the severe and critical groups from those that were less symptomatic is the excessive level of inflammation. In particular, elevated IL2R, IL6, TNF-alpha, and hsCRP in combination with suppressed lymphocyte counts have been seen in the critical group.97
Further, there is a growing consensus that IL2R and IL6 levels may confer the greatest advantage in predicting the severity of the disease. Nonetheless, it is my personal opinion that we should be looking at each individual’s case in-depth and considering the unique challenges and related nuances. Using such an approach demands that we better understand the various stages that the COVID-19 pathology may move through and when and how to apply certain interventions.
Unfortunately, the medical community does not yet have adequate resources to implement customized therapeutics at mass scale. For this reason, it will be your individual responsibility to understand your risks as thoroughly as possible and do your best to guide your medical professionals, should it be necessary, to select the right therapeutic approach, either for protection in place or as a response to an existing infection.
COVID-19 Stages: Step by Step
Stage I: Infection and Replication
Now that we have thoroughly described the basic process of immune response in the lungs, let’s take a deeper dive into how COVID-19 progresses, stage by stage, from infection to ARDS and beyond. Again, understanding where a patient is on the spectrum may be vital to providing the most appropriate support at the right time.
Given that SARS-CoV-2 is a positive-sense single-stranded RNA virus, it possesses the unique ability to use its genome as “messenger RNA” to hijack host cell ribosomal machinery. In other words, once inside a cell, it can use the cells own resources for producing proteins to generate its own proteins. So far in history, we’ve seen many such positive-sense RNA viruses, including hepacivirus C, West Nile, dengue, SARS, and MERS. The rhinoviruses that cause the common cold are also in this “privileged” category.
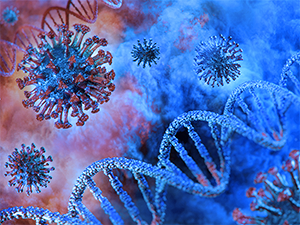
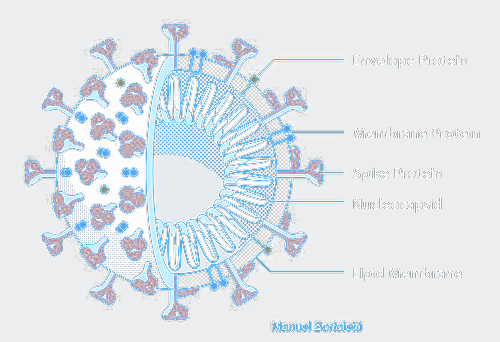
Like other members of the coronavirus family, SARS-CoV-2 has four structural proteins, including the spike (S) which confers the crown-like appearance, its envelope (E), membrane (M), and nucleocapsid (N), the latter of which carries the entire RNA genome. S, E, and M collectively constitute the total viral envelope. The spike protein, in particular, is responsible for allowing the virus to attach to and fuse with host cell membranes.
 Cellular Entry
Cellular Entry
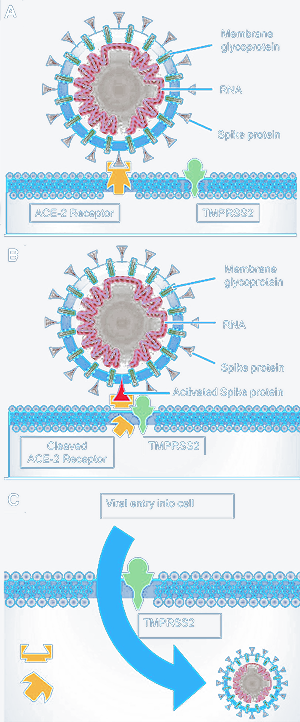
CoV-2 accomplishes this task by binding to angiotensin-converting enzyme 2 (ACE2) on cell membranes. This mechanism has been seen with the SARS-CoV virus from 2002-2003, though CoV-2 uses a different ACE2 site, considered to provide stronger binding affinity. This mechanism was elucidated already by January 2020 by a group of scientists stationed in both China and the United States using reverse genetics methods both independently and experimentally. ACE2 is found in abundance on epithelial cells throughout the respiratory tract, including trachea, bronchial, bronchial, and type I and II pneumocytes in the alveoli.
Once the spike protein has attached itself to a cell’s ACE2 receptor, a cathepsin, TMPRSS2, or another similar protease lodged in the cell’s membrane will cut open the spike. This exposes a peptide that inserts itself into the host cell membrane, thereby allowing fusion with the viral membrane. This in turn allows virion entry into the cell, where it may release its RNA genome.98
As you may have guessed, genetic expression of ACE2, TMPRSS2 and even cathepsins such as CTSB and CTSL are all relevant in determining an individual’s vulnerability to infection by SARS-CoV-2. In fact, a recent single-cell RNA-seq analysis has demonstrated that Asian males are likely to have much higher ACE2 expression, thereby increasing their risk for CoV-2 cell entry and inflammatory complications.99
There have been other reports that seem to confirm ACE2 variability in non-Asian populations as well. One such report describes a 33-year-old otherwise healthy German businessman that contracted COVID-19, only to suffer from a sore throat, chills, and myalgias with mild fever and productive cough. Literally 24 hours later (evening of the next day), he was feeling better and returned to work. One can’t but question this man’s genetic status for ACE2.100
Anecdotal evidence aside, we know very little regarding how well ACE2 polymorphisms correlate with COVID-19 disease severity. As stated elsewhere by other geneticists, a comprehensive analysis of quantitative trait loci along with potential coding variants in ACE2 will be needed in order to better understand implications. As I will describe at the end of this article, Transcend Genomics has performed an independent analysis for this protein, and a wide spectrum of binding affinities has been noted across hundreds of samples in our client database. It is, therefore, my opinion that certain changes in ACE2 protein configuration should confer protection against viral entry, at least to a degree. Nonetheless, binding affinity is only half the story, as receptor expression (and density) can easily overcome low binding affinities. That being said, I would therefore strongly advise caution when encountering “experts” claiming the ability to quantify your risk based on ACE2 SNPs. Population studies are not comprehensive enough to draw such conclusions, and statistics have proven consistently fallible.101
For example, one such recent genome study used data from the China Metabolic Analytics Project to identify 32 variants affecting the amino acid sequence of ACE2. Of these variants, ACE2 SNP rs2285666 was present in much higher frequency in Chinese populations than in American, African, or European. As a matter of fact, the homozygous mutation rate in males was much higher than females of the same Chinese population. In the same study, tissue expression data (GTEx) was analyzed and 11 additional variants were identified, all of which confer higher ACE2 expression in tissues. SNP rs4646127, in particular, had the highest frequency in Chinese and East Asian populations. Allele frequencies in European and American populations are also lower for this variant.
The conclusion was made that these polymorphisms may be associated with higher ACE2 expression, particularly in lung tissue, for East Asian populations. I feel obligated to reiterate — this conclusion was based on just over 10,000 people, far from being representative of global genetic variability at a global scale.102
Therefore, it should be abundantly clear that an insufficient amount of data has been collected to make statistically-driven conclusions about COVID-19 susceptibility in any population, globally. Therefore, I strongly advise caution when considering data coming out of consumer genetics-based services that rely on population-studies and allele frequencies to draw such conclusions. Until we have sequenced the genomes of at least 80% of the entire infected population across the full span of the pandemic, which is still evolving at the time of this article, we cannot be completely sure how ACE2 polymorphisms will behave. Coincidence abounds in statistical models and is resistant to control. Nonetheless, methodologies that look at the transcriptome along with protein binding affinities will fare much better in their ability to accurately predict outcomes.
Replication, Assembly, and Release
Once inside the cell, the CoV-2 virion’s membrane must be opened, most commonly through endosome-lysosome fusion and furin-mediated cleavage. Viral RNA may then be released outside the membrane and into the host cell’s cytosol. At that point, the RNA may be translated by the host cell ribosomes into fresh, new viral proteins. With increased viral load, the ribosomal “machinery” may be completely overwhelmed, dedicated in service to viral protein generation.
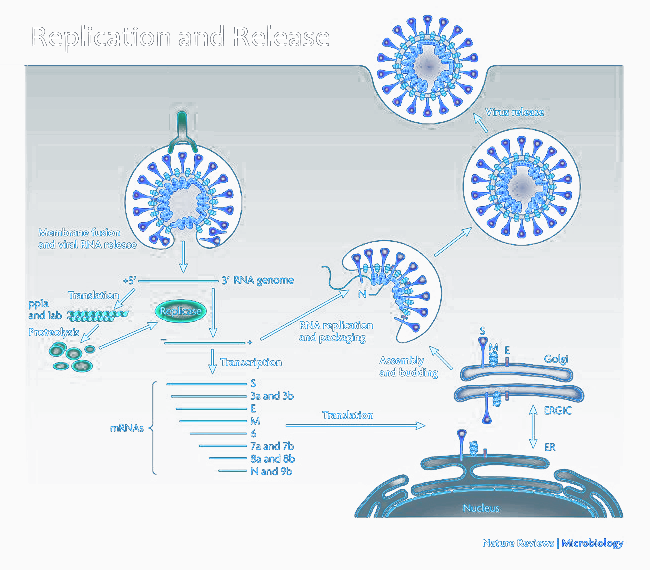
One of the first such proteins that are generated by the ribosome are small replicase-transcriptase complexes, which make their way into double-membraned, protective vesicles. In doing so, they place themselves farther away from viral-sensing mechanisms, thereby attempting to avoid recognition and replicate “in peace”.103
These “viral factories” begin translating viral mRNA by themselves, producing, piece by piece, the components of a complete coronavirus, including S, E, and M proteins. These proteins are, in turn, inserted into the endoplasmic reticulum where they make their way to the endoplasmic reticulum-Golgi compartment (ERGIC). This is their final stop, as the viral genomes encapsulated by the N protein bud into membranes on the ERGIC and combine with the E and M to form a membrane and envelope. The spikes are then incorporated and the fully-structured complex emerges into the cytosol as a mature virion.104105106107108
Once the virions have emerged, they are transported back to the cell surface by other vesicles. There, SARS-CoV-2 uses unique viroporin proteins (E and ORF3a) to form pores or “funnels” in the host cell’s membrane. These funnels serve as ion channels to propel virions out of the cell to the exterior, where they can proceed to infect other cells — and thus the cycle continues until immune factors bring the viral load down.
Virulence Factors
During the process of infection, SARS-CoV-2 will also produce at least three virulence factors which facilitates its ability to wreak further havoc:
- The first factor is Nsp1, which attaches to the 40S subunit of the ribosome, provoking it to degrade host cell mRNA. In the process, Nsp1 will also inhibit type-I interferon production, improving its chances for viral survival.109110
- Next, we have Nsp3c, which binds to cytosolic ADP-ribose, preventing it from participating in ADP-ribosylation, a vital post-translational process that plays a role in modulating immunological gene expression.111112
- ORF7a completes the circle of virulence by binding to and inhibiting activity of tetherin (BST2), an interferon-mediated membrane protein that “tethers” to virions, preventing them from leaving the cell.113114
Considering the nuances of infection and replication for SARS-CoV-2, it becomes clear that cellular resistance to attachment, infiltration, and replication is one of the deciding factors in how severe symptoms will be. Obviously, this particular strain of coronavirus is extremely adept at leveraging vulnerabilities in cellular defense, increasing its potential for higher viral load, especially in the lungs, where cells are rich with ACE2 receptor expression. In fact, analysis of throat swabs and bronchoalveolar lavage fluid (BALF) has shown up to 1,000 times more virions than was seen in SARS-CoV between 2002 and 2003. Nonetheless, it has been determined that BALF is far more accurate than throat swabs, due to the fact that the ACE2 and protease expression is much higher in the bronchi and alveoli.
Clinicians have been assigning Murray Scores to patients in order to help categorize the degree of acute lung injury. This score is based on the need for extracorporeal membrane oxygenation (EMCO) in severe acute respiratory failure. Not surprisingly, higher scores have been directly associated with viral load.115
Therefore, the earlier a high BALF viral load is detected, the greater the evidence an individual is more vulnerable to infection and high replication rate. Unfortunately, this translates to higher risk for excess inflammation and lung injury later in the progression of the disease.116
 Stage II: Inflammation and Lung Injury
Stage II: Inflammation and Lung Injury
Not everyone sustains lung injury from COVID-19, but nearly everyone experiences some degree of inflammation in the epithelium. As with infection and replication, there is also broad genetic variability in pro-inflammatory immune response, immunosuppression, and lung repair. Even though the replicase-transcriptase complexes locate themselves in double-membraned vesicles in an attempt to evade immune detection, nonetheless, as a viral load increases, the chances of triggering both innate and humoral immune responses increases.
Innate immunity is the first line of defense and is evolutionarily older and less dynamic than humoral response, which involves a large repertoire of learned antibodies. When it comes to viruses, a strong innate response is vital in the early stages of viral replication. As viral loads increase, however, this can lead to excessive production of pro-inflammatory cytokines.117 As mentioned earlier, airway epithelial cells can detect SARS-CoV-2 via their Toll-like receptors (TLRs). Activated TLRs initiate intracellular signaling cascades that induce an up-regulation in gene transcription for pro-inflammatory cytokines.
Multiple studies demonstrated that SARS-CoV (2002-2003) primarily infected airway and alveolar epithelial cells, vascular endothelial cells, and macrophages, and viral particles and viral genome were even detected directly in monocytes and lymphocytes. Therefore, the number of cells in the lower respiratory tract that could produce pro-inflammatory cytokines are numerous. In carefully balanced amounts, this will attract the proper immune response for neutralization of viruses. As a viral load increases, however, excess cytokine activity will effectively increase oxidative stress and damage healthy cells.118
Elevated levels of cytokines have been seen with other coronaviruses, including SARS and MERS. For example, increased IL1B, IL6, IL12, IFNγ, IP10, and MCP1 were associated with extensive pulmonary inflammation and lung damage in SARS, and MERS showed increased concentrations of interferon-gamma, TNF-alpha, IL15, and IL17.119 As expected, we see a similar cytokine profile with SARS-CoV-2, with IL1B, IFNγ, IP10, and MCP1 predominating.
In patients with insufficient viral control, viral loads outstrip immune-mediated containment, and we see a systemic attack on the body by the immune system, characterized by violent, destructive cytokine storms. Such storms, more often than not, lead to severe complications, organ failure, and death.120121
Hematopoietic Lineage and Immune Status
I’ve already stated my opinion regarding the correlation between immune cell senescence and high mortality rates in the elderly. We’ve also seen high mortality among immunocompromised younger adults, but the overall profile is identical: the immune system has shifted from balanced, tightly regulated production of erythrocytes and lymphocytes to skewed, uneven, “reactionary” adaptations that affect the entire blood cell lineage.
Clinicians have been busy looking at leukocytes, lymphocytes, and their subsets, yet there seems to be a paucity of consideration for the process of progenitor differentiation that gives rise to these separate families of blood cell types. All such cells, from red blood cells to basophils, neutrophils, eosinophils, monocytes / macrophages, natural killer cells, and T and B lymphocytes all come from a common parent: the multi-potential hematopoietic stem cell, also known as hemocytoblasts.
HSCs are produced in red bone marrow and differentiate, on average, into 500 billion blood cells per day. Depending on a vast number of environmental and internal triggers, the populations of all cell lineages must be tightly regulated. HSCs are limited in number and depend on perpetual self-renewal, a process that gradually declines with age. The two primary families of cells that arise out of HSCs are myeloid and lymphoid.
Once an HSC is differentiated into a myeloid progenitor, it can further morph into a megakaryocyte, erythrocyte (RBC), mast cell, or myeloblast (a.k.a. granulocyte). Granulocytes further differentiate into basophils, neutrophils, eosinophils, and monocytes, the latter of which may become a macrophage. From the lymphoid progenitor arise large granular lymphocytes (i.e. natural killer cells) and small lymphocytes. Small lymphocytes are well known for their ability to differentiate into T cells and B cells, the latter of which are responsible for antibody production.
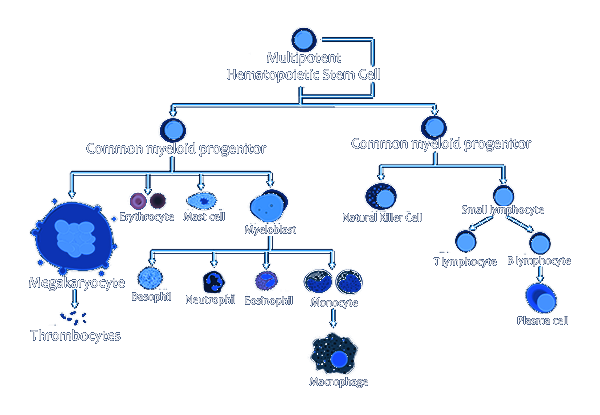
HSCs are highly mobile and, though self-renewal primarily happens in bone marrow, they are capable of passing the bone marrow barrier and travel through the blood either to other bones or make their way to the thymus. Those HSCs that settle in the thymus are generally predisposed to develop into T cells. Because of this mobility, it is possible to harvest HSCs from the blood, a process that is leveraged for bone marrow transplantation.
 Aging Thymus and Thymic Involution
Aging Thymus and Thymic Involution
The thymus is of particular importance in our consideration of age-related differences in immunological gene expression and their correlation with symptom severity. This small organ, located in front of the heart and behind the sternum, is the primary home for developing HSC-derived T cells. Such cells are considered to be highly self-tolerant and play a major role in overall protection from autoimmune-type attacks on the body’s own cells and organs.
Here, it’s important to note that the thymus is larger and more active in the period between birth and pre-adolescence and gradually wanes in both size and activity with age. In older individuals, thymocytes are gradually replaced by fat tissue, and T-cell development declines at an increasingly rapid rate with each decade.
This process is called thymic involution and begins as early as one year after birth — an “atrophy” that is spurred on during adolescence by the appearance of sex hormones. As a matter of fact, the thymus is shown to increase in size post-castration. However, sex hormones are not the only factors that can influence thymus size and function. Any severe illness, especially one that results in immunodeficiency such as HIV, can provoke thymic involution. That being said, regardless of infection status, once thymic involution occurs, it is difficult if not impossible to reverse. Even after bone marrow transplant, which should replenish the HSC pool, patients over the age of 40 may still be unable to regenerate the thymic compartments for naive T-cell maturation.122
In coronavirus infection, an individual’s capability to mount a robust antiviral response depends on the diversity in receptors on naïve T-cells. These naive T lymphocytes are highly tolerant to “self-antigens” (i.e. proteins that attack one’s own body) and quite agile in their ability to respond to foreign invaders. When we say their receptors are “diverse”, we mean that they are poised to respond to pathogenic threats under a wide array of contexts. As thymic involution progresses, the number of diverse receptor types on naïve T cells declines. Already by the age of 65, T-cell receptor diversity has completely gone off a cliff. It is for this reason that the elderly succumb more easily to infection — they have lost their immune resilience and have a limited T cell receptor repertoire.123124
To make matters worse, there are other changes in an aging thymus that can complicate antiviral immunity. Normally, any developing T cells that are hyper-reactive to self-antigens are immediately given the death sentence. Unfortunately, non-self, pathogenic antigens can make their way into thymocytes and turn everything upside down. Instead of eliminating the self-hyper-reactive T cells, in the presence of foreign antigen, the pathogen-targeted T-cells are removed instead. In coronavirus infections that become systemic, viral loads are soaring, and active T lymphocyte counts plummet, partially as a result of virally-amplified thymic involution. To add insult to injury, the self-reactive T lymphocytes can make their way into circulation, inducing increased autoimmune-mediated inflammation elsewhere in the body, including the lungs.125
In this scenario, we see an increasing trend for myeloid progenitors to pick up the slack in immune response and up-regulate granulocyte differentiation. This appears on blood draws as increased neutrophils, monocytes, and macrophages, possibly with elevations in basophils and eosinophils as well, depending on immunological disposition. As one would expect, because red blood cells are in the same lineage as granulocytes, at the height of infection, there is also potential for altered erythropoiesis, especially if there are concomitant issues in the kidneys (a frequent complication of late-stage COVID-19 illness), which are responsible for secreting EPO (erythropoietin). EPO is yet another cytokine whose expression is up-regulated in the kidney in response to hypoxia. It stimulates the production of red blood cells from HSCs in bone marrow — a response that is significantly hindered in severe illness where demand for HSC differentiation has outstripped their ability for self-renewal, which declines with age as well.
At the onset of SARS-CoV-2 infection, acute elevations in lymphocytes have been noted. However, in individuals that progress to more severe stages, lymphocyte counts are suppressed and we see a predominance of monocytic/macrophagic activity. For this reason, higher levels of GM-CSF have been observed in the blood serum of patients in late-stage COVID-19 illness. GM-CSF is a cytokine secreted by macrophages that is responsible for stimulating HSCs to produce granulocytes (neutrophils, eosinophils, and basophils) and monocytes. Monocytes, in turn, make their way into the bloodstream where they circulate for 1-3 days before eventually migrating into tissues and differentiating into macrophages and dendritic cells, thereby magnifying the inflammatory cascade that further amplifies macrophage activity.
This problem is further complicated in the lungs where there are pre-existing alveolar macrophages. These lung macrophages secrete GM-CSF to stimulate their own proliferation (via monocyte differentiation), and as viral loads increase, there is chronic macrophage accumulation. As I will describe in more detail below, macrophages not only produce pro-inflammatory cytokines, but can be directly infected by SARS-CoV-2, given the presence of ACE2 receptors on their membrane. As such, alveolar macrophages pose significant inflammatory potential in an environment with ever-increasing viral loads.
Spleen: Erythrocyte and Iron Regulator
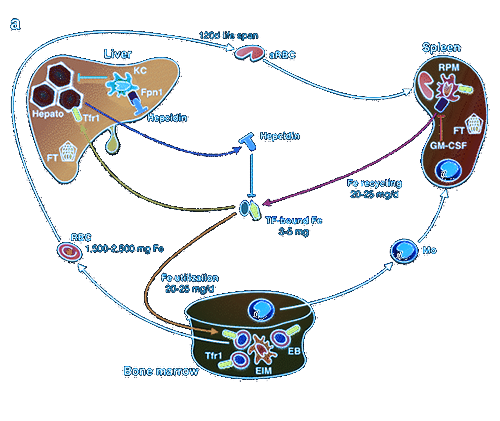
The last component of the immune system that determines an individual’s overall immunological vulnerability is the spleen. The spleen is extraordinarily relevant in COVID-19, for reasons that will become more apparent further in this article. Suffice it to say now that SARS-CoV-2 causes massive changes to blood cell homeostasis, which may include hemolysis of red blood cells and excess monocyte/macrophage activity. Given that the spleen plays a primary role in removing old or ruptured red blood cells as well as recycling iron, it’s clear that any disturbance to its internal processes could be detrimental to the body as a whole. As vascular permeability increases in the lung, there is increasing risk for red blood cell translocation, which can result in rupture and release of hemoglobin extravascularly. As I have already stated, this is not a prominent characteristic of COVID-19, but could easily play a role in later stages of the disease. In scenarios of increased hemolysis, the spleen will be under considerably higher stress, degrading cell-free hemoglobin and metabolizing heme to bilirubin, which is in turn removed by the liver.
Further, the spleen both synthesizes antibodies and removes antibody-coated bacteria and red blood cells via blood and lymph node circulation. Moreover, half of the body’s monocytes, differentiated from HSCs in bone marrow, make their way to the spleen — therefore, any disturbance in the myeloid lineage can cause changes or obstructions in proper spleen function, leading to skewed blood levels of various myeloid cells.126
Inflammation Caused by Rapid Viral Replication and Cellular Damage
As the inflammatory cascade intensifies, due to both rapid viral replication as well as “runaway” macrophages, we start to see massive damage to the lung epithelium which leads to leakage in the arteries and capillaries surrounding the alveolar sacs. This provokes progressively more intense “cell danger” signals and aggressive, even violent release of more cytokines and chemokines. As alveolar epithelial cells are damaged, an increasing number of cytotoxic (CD8+) T cells are recruited to attempt to destroy the virions, which contributes to the T-cell “exhaustion” and thymic involution described above. All the while, excess inflammation is invoking apoptosis (i.e. cell death) in type I pneumocytes.127 Over time, the situation becomes so dire, that the surface area for gas exchange on type I pneumocytes decreases, and oxygen can no longer be effectively diffused for binding with deoxygenated hemoglobin in red blood cells.
Throughout this process, virions continue to stimulate Toll-like receptors on the surfaces of alveolar macrophages. This leads to actin polymerization in the macrophage, resulting in a suppression of integrin expression and a deactivation of TGF-beta, both of which are responsible for maintaining macrophage attachment to the alveolar epithelial cells. As a result, the alveolar macrophages detach from the AECs, primed for attack, secreting pro-inflammatory TNF-alpha and IL6. As you likely recall, elevated IL6 (along with IL2R) is one of the primary defining indicators for severe ARDS in COVID-19. Once mobilized, circulating interferon-gamma increases NADPH oxidase’s affinity for NADPH in macrophages, raising potential for “respiratory burst”, enhancing TNF-alpha secretion. In this way, SARS-CoV-2 provokes alveolar epithelial cells to liberate their macrophages and induce an aggressive pro-inflammatory response.128
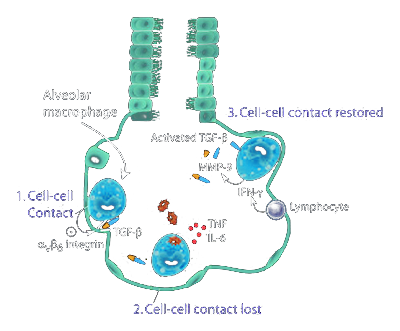 Normally, alveolar macrophages (AMs) are kept in a dormant state, due to the high sensitivity of type I and II pneumocytes to damage. In this state, AMs suppress T and B cell activity, given such immune cells may lack target specificity. In this way, AMs keep the innate and humoral immune systems from provoking collateral damage in AECs and invoking uncontrolled inflammation. Dormant AMs further maintain balance by secreting nitric oxide, prostaglandins, IL4 and IL10, and TGF-beta, the latter of which is inhibited during their mobilization.129130131132133
Normally, alveolar macrophages (AMs) are kept in a dormant state, due to the high sensitivity of type I and II pneumocytes to damage. In this state, AMs suppress T and B cell activity, given such immune cells may lack target specificity. In this way, AMs keep the innate and humoral immune systems from provoking collateral damage in AECs and invoking uncontrolled inflammation. Dormant AMs further maintain balance by secreting nitric oxide, prostaglandins, IL4 and IL10, and TGF-beta, the latter of which is inhibited during their mobilization.129130131132133
Nitric oxide, produced by inducible NOS (i.e. NOS2) by alveolar macrophages is an important player in the anti-inflammatory milieu. It primarily acts through an auto-regulatory feedback loop that involves bacterial/viral stimulation of pro-inflammatory cytokines, cytokine stimulation of NO production, and resulting NO down-regulation of cytokine expression. As a matter of fact, NO inhibits the GM-CSF mediated maturation of dendritic cells. In existing dendritic cells, NO further increases their capacity to internalize viral antigens, directly at sites of inflammation, improving antigen-specific responses.
It is explicitly in this way that increased hyper-reactivity to antigens provokes elevated levels of nitric oxide in the pathology of asthma. The greater the antigen “load”, the more regulatory nitric oxide will be present to attempt to contain cytokine expression. For this reason, NO is often measured in the breath of asthmatics to determine the degree of disease severity. Clearly, nitric oxide balance is a very delicate matter that can only be understood in the context of individual response to antigens and ability for nitric oxide production — as well as pre-existing conditions which could provoke its accumulation.134
Though science has been challenged to thoroughly delineate the origins of asthma and similar pulmonary disorders, it is nonetheless of interest that alveolar macrophages secrete anti-inflammatory cytokine IL4 in order to maintain immune balance in the lung epithelium. This is an extremely important point, because IL4 plays a key role in promoting naive T cells to differentiate into mature T-helper 2 type cells. This is a desirable outcome in an environment that is prone to excess inflammation, but in those individuals that are hyper-reactive to antigens, there can be an overabundance of IL4 expression.135136
This can become problematic in situations of acute lung insult, given that IL4 can have paradoxical effects on alveolar macrophage homeostasis. On the one hand, it promotes AM phagocytosis of virions. On the other hand, it has also been shown to inhibit AM production of prostaglandin PGE2, which is vital for the enhancement of peripheral blood lymphocyte IL10 transcription and protein production. IL10 is vital in such contexts for the promotion of immune tolerance and mitigation of inflammatory processes. Further, PGE2 has also been shown to deactivate macrophages and T-cells, a desirable outcome in a hyper-inflammatory state. Therefore, IL4 inhibition of PGE2 and, in turn IL10, can result in amplification of the inflammatory cascade.
In such contexts, with lower IL10 secretion, there will be less inhibition of pro-inflammatory TNF-alpha, IFN-gamma, possibly resulting in excessive T cells and NK cells but, more importantly, higher proliferation rates for alveolar macrophages, themselves. In this way, what would otherwise be a balanced biofeedback mechanism to maintain peace and quiet in the lung epithelium can become the perfect storm for Th2-mediated inflammatory collateral damage.
To reiterate, alveolar macrophage secretion of immune-suppressive IL10 generally only happens in the dormant state, something that is not seen under excessive viral load. Nonetheless, for those AMs that have not yet been provoked to action, Th2-type immune dysregulation, as seen in models of asthma, could increase vulnerability to even higher levels of inflammation, both from activated AMs as well as from those producing excessive IL4.137 Nonetheless, elevated IL10 has been noted in a large number of COVID-19 ARDS cases and has generally been associated with potential for “immune paralysis”. Clearly, such broad generalizations cannot be made, especially without knowledge of the patient’s pre-existing immune status. In fact, I have not yet seen a thorough explanation of what the implications of excess IL10 are and, as should be quite obvious by now, I believe cytokine levels can mean different things for different people.
In the case of COVID-19, there is the additional possibility (not yet posited by scientists to date) that SARS-CoV-2 could potentially modulate alveolar macrophage IL10 expression in some manner to facilitate infection. As a matter of fact, other microbes such as Yersinia enterocolitica have already demonstrated an ability to release virulence factors which can induce IL10 via TLR2 and CD14 TLR4 receptors resulting in complete suppression of interferon-gamma and TNF-alpha — a decidedly positive anti-inflammatory effect but devastating for antimicrobial resistance.138
In fact, this type of dysregulation in AECs appears to be present in COVID-19 ARDS as well, characterized by hyper-activation of alveolar macrophages and IL10-mediated suppression of antiviral immune response. Without returning mobilized macrophages to their dormant state, gas exchange across type I pneumocyte membranes can be compromised due to both excess inflammation and pneumocyte damage. Normally, activated lymphocytes achieve this by secreting IFN-gamma which in turn provokes macrophages to produce matrix metalloproteinase MMP9 — MMP9 then activates latent TGF-beta which re-instates the integrins on AECs, thereby pulling macrophages back into a resting state. As disease severity increases, IL10 mediated lymphocyte suppression becomes more prominent, and there is generally less IFN-gamma in the lung epithelium, lower MMP9, and a loss of control over macrophage state. This is where we see macrophage accumulation, excess inflammation, and, ultimately, lung tissue remodeling.139
For this reason, it is common to see an increased neutrophil to lymphocyte ratio (NLR) alongside T lymphopenia in the most severe COVID-19 cases. Not surprisingly, this includes a notable decrease in CD4+ T cells. This is in part due to thymic involution, as described above, but HSC differentiation is also likely to be skewed, with a preference for myeloid-lineage cell types, especially erythrocytes — which may be direly needed as hypoxia/oxygen deprivation elevates.140141142
Pyroptosis in Macrophages and Lymphocytes
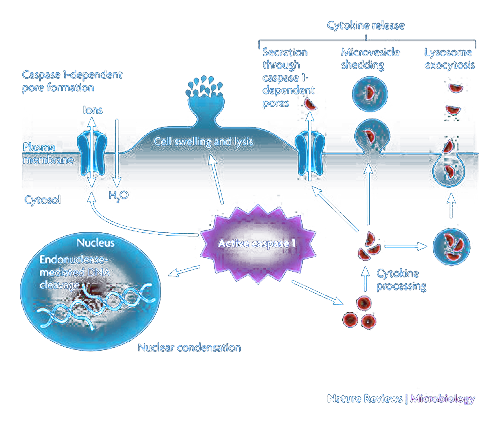
Another reason for widespread lymphocyte suppression is due to the ability of SARS-CoV-2 to induce T-cell “death” via inflammasome activation, also known as pyroptosis.143 Once SARS-CoV-2 has infected a cell, it can activate NOD-like receptor protein complexes (NLRPs), also known as inflammasomes.144145
Inflammasomes consist of three proteins — a NOD-like receptor, an adapter (ASC), and the effector molecule pro-caspase-1 (CASP1). Once the inflammasome has been “turned on” by contact with virion proteins, CASP1 is cleaved, which in turn mediates the cleavage of pro-inflammatory cytokines IL1B and IL18, activating them and preparing them for secretion. Activated CASP1 has also been shown to initiate the cleavage of gasdermin D (GSDMD), which is the primary trigger for pyroptosis.
Pyroptosis occurs due to further cleavage of GSDMD to produce two separate domains (N and C). The N domain fuses with the lymphocyte’s or macrophage’s membrane from the inside, creating a transmembrane pore. IL1B and IL18 are secreted via that pore, along with other contents of the cell. This essentially disrupts the cell’s ionic gradient, causing a drastic increase in osmotic pressure and subsequent influx of water. This causes the cell to swell and burst.146
What is important to understand here is that inflammasome activation is not inherently an undesirable event. In fact, IL1B secretion is indispensable for protection against viral infection. In mice that have had their IL1B gene nullified, viral loads increase while inflammation decreases. This underscores the importance of a balance between antiviral activity and excess inflammation. Clearly, one cannot exist without the other.147
IL18, in particular, plays a vital role in keeping the antiviral side of that balance by potently inducing interferon-gamma expression, in concert with IL12. This in turn provokes natural killer cells to action. Taken together, there is ample evidence that IL1B and IL18 play crucial roles in antiviral immunity and, in the early stages of viral infection, are indispensable to containing viral load. Nonetheless, they are both highly pro-inflammatory, and uncontrolled IL1B secretion is tightly correlated with the severity of clinical symptoms, especially in patients with auto-immune disorders.
So any therapy that would mitigate over-expression of the cytokines would also be expected to increase viral load and possibly complicate outcomes. The question is less about how to suppress inflammation or prevent invocation of inflammasomes such as NLRP3 but, rather, to more thoroughly understand individual genetics for immune expression, from HSCs, thymus, and spleen to dynamics for alveolar macrophage mobilization and containment. We will be discussing in detail multiple strategies to accomplish this goal further along in this article, but first let’s go deeper into the details of how the more severe stages of this disease progress.
 RAS Dysregulation
RAS Dysregulation
There has been much discussion during the COVID-19 crisis surrounding ACE2 receptors, particularly in the lungs, but it has mostly been relegated to SARS-CoV-2 cell entry. A more in-depth consideration of the renin-angiotensin system (RAS) is required to fully appreciate the matter in which this virus can affect the cardiovascular and excretory systems, particularly with respect to the kidneys.
The RAS system hormonally regulates blood pressure and systemic vascular resistance through its effects on fluid and electrolyte concentrations. Whenever there is a drop in blood pressure, the kidneys respond by harvesting circulating precursor prorenin, converting it to renin in the juxtaglomerular cells, and then secreting renin back into the bloodstream. At this point, in order for renin to be able to promote an increase of blood pressure, there must be a sufficient level of angiotensinogen present, the secretion of which is provoked in the liver by corticosteroid (e.g. cortisol), estrogens, and thyroid.
Once renin is in the bloodstream, it can take angiotensinogen and convert it to angiotensin I, which is subsequently picked up by the angiotensin-converting enzyme (ACE) — residing predominantly in the lungs — where it is converted to the more potent and active peptide, angiotensin II.
As I will describe in more detail below, angiotensin II (AngII) has a widely varied repertoire of effects, some of which are not well studied. AngII’s most prominent effect is the widespread constriction of blood vessels, thereby increasing blood pressure. AngII has also been shown to stimulate the excretion of the hormone aldosterone in the adrenal cortex, which in turn causes renal tubules in the kidney to reabsorb sodium. In the process, a significant amount of water is drawn back into the blood, and potassium is excreted. In this way, extracellular fluid throughout the body is elevated, resulting in increased blood pressure.
In some individuals, the RAS system can be overactive. This can be of genetic origin or as a result of unhealthy lifestyle adaptations. SARS-CoV-2, in part via its interrelations with ACE2, can also change dynamics in this system, potentially leading to elevated blood pressure. The higher blood pressure goes, the greater the potential for damage to the kidneys, further decreasing their ability to regulate fluid dynamics in the body and maintain proper erythropoietin levels.
One of the ways that the body regulates ACE expression is via its counterpart, ACE2, which is also found attached to cell membranes in the lungs, arteries, heart, kidney, and intestines. ACE2 effectively lowers blood pressure by degrading AngII, via hydrolysis, to angiotensin (1-7), a vasodilatory septapeptide. Unfortunately, SARS-CoV-2 uses ACE2 specifically to gain entry to cells leading to ACE2 “shedding”. Therefore, blocking or inhibiting ACE2, for the purpose of blocking viral entry, carries a very clear risk of increased hypertension with potentially fatal consequences. As a matter of fact, ACE2-mediated production of angiotensin (1-7) has shown a decidedly lung-protective effect in models of virus-induced injury. Given that SARS-CoV-2 causes a sharp drop in ACE2 expression by promoting receptor internalization and degradation, loss of ACE2 is quite likely one of the ways in which this virus causes such extensive lung damage.148149
Most of the hypertension drugs used in the world today manipulate the RAS system in one way or another, either by blocking receptors for AngII with ARBs (angiotensin receptor blockers) or by inhibiting the conversion of AngI to AngII with ACE inhibitors. ACE2 agonists or enhancers have been considered for therapeutic purposes as well.150151
It should, therefore, be of significant interest that both ARBs and ACE inhibitors have demonstrated the capability to up-regulate ACE2 expression, though the knee-jerk reflex among the public at large has been to presume that this will increase the “attack surface” for coronavirus entry in the cells.152153
However, in reality, it is not that straightforward. In fact, multiple studies have implied a significant increase in lung injury resulting from SARS-CoV-mediated shedding of ACE2.154155156157 As ACE2 is progressively lost, RAS-system dysfunction proceeds, further enhancing inflammation, vascular permeability, neutrophil accumulation, and overall diminished lung function.158
ACE2 shedding occurs not only as a result of the release of enzymatically active soluble ACE2 (sACE2) via the action of disintegrin and metalloprotease 17 (ADAM17), but can also be induced directly by pro-inflammatory cytokines such as IL1B and TNF-alpha — the latter of which is released upon NLRP3 inflammasome activation.159160161 Therefore, contrary to expectation, many medical professionals continue to recommend the use of ARBs and ACE inhibitors in the course of COVID-19 disease. In a meta-analysis published on July 11, 2012, ACE inhibitors were associated with a significant 34% reduction in pneumonia risk, particularly in those with pre-existing risk for stroke and heart failure.162
As inflammation becomes progressively worse, partly due to loss of ACE2 function, there is increasing potential for damage to the lungs. Such damage invokes cellular regeneration processes that, under normal conditions, maintain the integrity of lung tissues. Under continued pressure, however, activation of cellular adhesion proteins such as beta-catenin occur. Beta-catenin activation can lead to the ectopic differentiation of type II pneumocytes, causing goblet cell hyperplasia and air-space enlargement.
Lung injury further provokes expression of TGF-beta, a growth factor which, if over-activated, can lead to pulmonary fibrosis163 by enhancing mesenchymal cell proliferation and synthesis of the ECM (extracellular matrix).164 As mentioned earlier, type II pneumocytes, along with other specific stem cells, function as progenitors for type I pneumocytes, the latter of which is responsible for gas exchange in alveoli. After lung injury, type II pneumocytes have shown the capability for self-renewal, giving rise to new type I AECs.165166
However, with higher viral loads, we will see less effective containment of lung injury, leading to an excess of progenitor cells with hyperplasia (i.e. overgrowth) of type II pneumocytes. This is accompanied by increased deposits of fibrin and collagen in alveolar spaces, resulting in a loss of crucial surface area for optimal gas exchange.167 This reduction in gas exchange is accompanied by severe damage to type I pneumocytes, leading to their death by apoptosis.
One of the reasons that we see elevated IL6 in severe ARDS is because of the role it plays in lung cell regeneration, similar to such processes in the liver. In fact, HGF (hepatic growth factor) levels increase in the liver in parallel with IL6.168169 Similarly, IL6 has a proliferative effect on pulmonary fibroblasts. In small amounts, it can help regenerate lung tissue, but with persistent, acute damage, it provokes hyperplasia and loss of type I pneumocytes. Therefore, the magnitude of injury to the alveolar epithelial barrier is tightly correlated with the severity of pneumocyte hyperplasia.170
It has been suggested that HGF could be used as a means to regenerate injured lung tissue without the side effect of type II pneumocyte hyperplasia. In fact, it would appear that the lung may actually be a direct source of HGF when liver production capacity has declined for any reason.171 At least one study has demonstrated that recombinant HGF, administered 2 weeks post-onset of pulmonary fibrosis, decreases collagen levels in lung interstitia. This may indicate that HGF could very well be capable of halting or reversing the progression of fibrosis.172
Thyroid hormone and acute lung injury
Another factor in the development of ARDS that is not often talked about is the effect that thyroid hormone has on type I pneumocytes. It is long been known that prenatal administration of glucocorticoids can effectively promote type II pneumocyte maturation, thereby mitigating respiratory distress in infants. However, thyroid hormone binding to its receptor in the lung can also directly affect maturation of type I pneumocytes. For this reason, thyroid medication such as Cytomel, may be effective at mitigating respiratory distress when glucocorticoids have proven ineffective. Synthroid (i.e. synthetic thyroxine), on the other hand, may actually have the opposite effect — which I will discuss further below in the section on BiopathFx predictions.173
Stage III: RBC and Hemoglobin Disruptions
The next stage in COVID-19 progression is, in fact, hypothetical. We have seen pink exudate in the lungs of ARDS patients, and some have postulated there may be a degree of hemolysis occurring. I have seen some self-proclaimed experts even citing hemolysis as the primary driver behind SARS-CoV-2 induced ARDS.
Here, I would like to reiterate — frank hemolysis or derangements in the oxygen-carrying capacity of hemoglobin have not been demonstrated in COVID-19. The conclusions surrounding altered hemodynamics, as I will explain, are based on some basically flawed premises derived from at least one questionable scientific paper. Nonetheless, it is clear that some degree of disruption in hemodynamics is quite possible in later stages of this disease. Therefore, an in-depth discussion of the risks is in order.
In reality, hemolysis and resulting cell-free hemoglobin and heme, would not even make an appearance until there has already been sufficient alveolar and vascular damage, allowing erythrocytes (RBCs) to permeate into lung tissue. Symptoms of respiratory distress would present long before this happens.
Erythrocyte Rupture (Hemolysis)
As ACE2 receptors are progressively down-regulated by SARS-CoV-infection in alveolar epithelial cells, they are simultaneously shed in the vascular epithelium, particularly the capillaries enveloping the alveoli. This leads to increased AngII levels in blood serum, elevated blood pressure, and higher AngII-mediated inflammatory damage directly to the vasculature. Over time, this results in vascular permeability, and red blood cells may be permitted to escape circulation. In the process, because of the tight spaces they must pass in the vascular endothelium, they can be squeezed and deformed. The higher blood pressure is, the more force there will be pushing red blood cells through the vasculature. Ultimately, this can result in rupture, and release of hemoglobin directly into the extracellular space.
This cell-free hemoglobin can be driven to a number of fates, though the most damaging form is actually the oxygen-carrying form known as “oxyhemoglobin”. This reduced ferrous oxyhemoglobin is highly reactive with nitric oxide and is capable of rapidly scavenging available NO to form methemoglobin and nitrate.174175
Once in the methemoglobin form (i.e ferric hemoglobin), it can further be catalyzed to free heme and ferric iron, which can sustain further damages, though it should be noted that in order to form the methemoglobin, it is necessary to deplete nitric oxide, thereby worsening vasoconstriction, hypertension, and increasing vulnerability for infection. Therefore, the original and most deadly reaction starts with the reduced ferrous-form oxyhemoglobin — a factor that will become more pertinent as we discuss therapeutic strategies aimed at preventing methemoglobin formation.
Under normal conditions, the body scavenges cell-free hemoglobin by provoking the liver to produce haptoglobin. Free heme, in turn, is scavenged by liver-derived hemopexin. In this way, redox-active molecules are prevented from producing cell-damaging reactions. Unfortunately, with higher degrees of hemolysis — which, to re-iterate, have not been demonstrated in COVID-19 — the scavenging systems are completely saturated by excess cell-free hemoglobin, and that hemoglobin rapidly accumulates, both inside the vasculature and outside in tissues and organs.176
This process may be summarized in the figure above, which demonstrates how hemolysis releases cell-free hemoglobin, thereby inducing vascular injury. The more hemoglobin is kept in its reduced oxyhemoglobin state, the greater its potential will be for reactivity with nitric oxide which, in turn, inhibits NO signaling further. This is particularly a problem in subendothelial space (i.e. when hemoglobin has escaped circulation and entered tissues). In this situation, reductive recycling back to the ferrous state (e.g. with ascorbic acid or other reducing agents) will enhance the pathology of nitric oxide depletion. Nonetheless, damage occurs in multiple metabolic directions, as the methemoglobin produced from oxyhemoglobin reactivity with NO may further oxidize to the ferryl state and drive Fenton and peroxidase oxidative cellular damage. These reactions are particularly lethal to kidney cells and will result in acute kidney injury.
Some methemoglobin may also separate into free heme and ferric iron, both of which are capable of inducing comprehensive inflammatory injury via their activation of macrophages in monocytes. Without proper haptoglobin or hemopexin expression, these dangerous reactions will become more prevalent and with amplified hemolytic activity, the less haptoglobin/hemopexin output we will see. Again, this kind of Hp / Hx exhaustion is not likely in COVID-19 ARDS. Therefore, some of the proposed therapies using reducing agents may work — perhaps sheerly due to containable, low-level hemolysis and resulting cell-free hemoglobin.
Once again, it’s important to underscore that acute hemolysis has not been observed in COVID-19. On the contrary, it is more commonly the result of blood transfusion or bleeding disorders. As a matter of fact, the above-noted effects have been duplicated in studies observing transfusions with whole blood. Given the increased fragility of red blood cells in aged blood, there is a higher potential for intravascular hemolysis, resulting in an accumulation of reduced ferrous oxyhemoglobin. In order to resolve this problem, recombinant haptoglobin has been included with such transfusions.177
Such haptoglobin-saturated transfusion has been demonstrated to significantly mitigate the vasoconstrictive effects seen with cell-free hemoglobin. Nonetheless, regardless of haptoglobin saturation, oxyhemoglobin reactivity with nitric oxide can still be an issue, apparently due to haptoglobin’s ability to increase oxyhemoglobin’s half-life. This leads to questions about how haptoglobin is capable of modulating vasodilation and/or NO signaling. This can probably be attributed to the fact that NO signaling normally occurs in a cell-free zone between the endothelium and red blood cells, a zone that is disturbed in models of excess vascular permeability. Therefore, the viability of using either redox agents and/or haptoglobin for the purpose of containing damage from cell-free hemoglobin may be in question — specifically in those contexts.
That being said, two of the primary biomarkers for the quantification of hemolysis are serum haptoglobin and LDH (lactate dehydrogenase). LDH levels have been normal in the majority of COVID-19 patients, with the exception of those in the ICU, where we begin to see elevations. This does not indicate a direct correlation with hemolysis. Quite the opposite, really — LDH increases with any and all tissue damage and is particularly high in those suffering from interstitial lung disease with hypoxia. There have been zero indications of elevated serum haptoglobin in COVID-19 patients with ARDS. Therefore, we can make no firm conclusions about its pathology at later stages of disease.178179
Nonetheless, since there are experts in the field promoting the use of reducing agents to keep cell-free hemoglobin in a form “bindable” by haptoglobin, I would like to add an extremely important nuance here. As we have already discussed, increased oxidative stress in the lung can result in higher expression of superoxide dismutase, which neutralizes superoxide, separating it into oxygen and hydrogen peroxide. Therefore, higher levels of oxidative stress in more severe stages of ARDS may also allow the accumulation of hydrogen peroxide in the lung. In the presence of cell-free hemoglobin, this excess hydrogen peroxide could produce a Fenton reaction with reduced ferrous oxyhemoglobin to form hydroxyl radicals, a problem — once again — that would be exacerbated by any reducing agents keeping cell-free hemoglobin in its ferrous state.
In fact, vasopressor (i.e. hypertension) effects of cell-free hemoglobin have been shown to directly correlate with NO scavenging reaction rates and with molecular size.180181 The more oxygenated cell-free hemoglobin there is, the more acute the hypertensive response will be. This vasopressor response becomes particularly problematic in the lungs, where cell-free hemoglobin is known to provoke pulmonary hypertension and oxidative injury.182183
The question arises — where is hemolysis a greater concern; in the vascular system or in the airways? This is an important consideration when considering whether anti-hemoglobin or anti-heme strategies are administered intravascularly or directly into the airways.184
Intravenously administered red cell antioxidants, for example, may be too rapidly metabolized in the airway space before they are able to effectively counter vascular permeability and other inflammatory effects of cell-free hemoglobin and heme. Cell-free heme, in particular, is potently pro-oxidant due to its hydrophobic nature, capable of promoting lipid peroxidation and even stimulating inflammation directly via TLR4.185186187
Oxygenated hemoglobin (i.e. ferrous oxyhemoglobin) is capable of scavenging nitric oxide 1,000 times faster than its equivalent inside red blood cells, so even low levels of hemolysis could be quite problematic, if encountered during COVID-19.188 Liver haptoglobin binds to oxyhemoglobin, thereby preventing it from reacting with nitric oxide. Though the majority comes from the liver, it may also be synthesized directly in the lung but, as I will describe below, its supply sharply declines as hemolysis progresses.189 In fact, those individuals most likely to survive sepsis are those with higher haptoglobin levels, but this does not necessarily mean that they have higher genetic expression or altered antioxidant status. Rather, it’s an indication of their “cytokine load” and degree of hemolysis, which is inversely correlated with haptoglobin levels.190
That being said, RBCs will not hemolyze directly in the alveolar compartment until there is widespread vascular permeability. This is because RBCs are far too large to cross an even mildly permeable endothelium. There must be excessive damage to the vasculature in order for a red blood cell to appear in the alveoli, and by the time that has happened, it has most likely ruptured in the process. So if there are low to zero levels of hemoglobin in bronchoalveolar lavage (BALF), it is likely a safe assumption that the ceiling for vascular permeability has not yet been exceeded. Further, it’s important to consider that blood in BALF does not equate to “cell-free hemoglobin in BALF”. Red blood cells can (and do) reach the extravascular space without rupture. In fact, one of the reasons that encapsulated hemoglobin is less toxic is because the RBCs themselves contain antioxidants such as glutathione, catalase, and peroxiredoxin-2, all of which prevent oxidative tissue injury. RBCs in the alveolar space could even be considered to be protective, in this sense.191
One of the reasons that haptoglobin levels decline with increasing severity of hemolysis is because of liver stress from excess bilirubin. Once cell-free hemoglobin is bound to haptoglobin, it is taken up by macrophages in a CD-165 receptor-mediated manner and converted to bilirubin by heme-oxygenase-1 (HMOX1). Bilirubin accumulation then increases liver-mediated glucuronidation, quickly consuming available glucuronic acid stores systemically. This results in a lower ability to glucuronidate other substrates such as sex hormones or NSAIDs (e.g. acetaminophen), thereby inducing liver injury. This has widespread effects on not only haptoglobin production but clearance of toxins from the bloodstream as well. The kidneys, also stressed from hypertension and toxin accumulation will eventually fail as well in this scenario.192193194
Clearly, the reason inhaled nitric oxide therapy has provoked unwanted effects in specifically “hemolytic” disorders is because of haptoglobin depletion, increased oxyhemoglobin, uncontained hemolysis, and chronic, prolonged scavenging of any added NO to that system. This is precisely the mechanism by which we see massive NO consumption in sickle-cell disease. I must strongly advise any clinician reading this article to consider the implications of using redox molecules to reduce methemoglobin to ferrous oxyhemoglobin in the presence of hemolysis. This may only be effective at the early stages characterized by slower accumulation of oxyhemoglobin. I will be discussing this topic in more detail in the section below covering ascorbic acid. Suffice it to say now that we do not want elevated cell-free oxyhemoglobin in the absence of haptoglobin and/or hemopexin. This will only further drive down endothelial nitric oxide levels, exacerbate hypertension, and pour oil on the fire of hemolysis.195196
In fact, blood transfusions with HBOCs (hemoglobin-based oxygen carriers), used to resolve hypoxia/oxygen starvation, will categorically fail for the reasons I stated above if not combined with recombinant haptoglobin. No amount of antioxidants in the world will rescue a body suffering from nitric oxide deficit and haptoglobin suppression. How relevant this dynamic is in COVID-19 is a matter of debate at this time.
Disappearing Act: Haptoglobin / Hemopexin
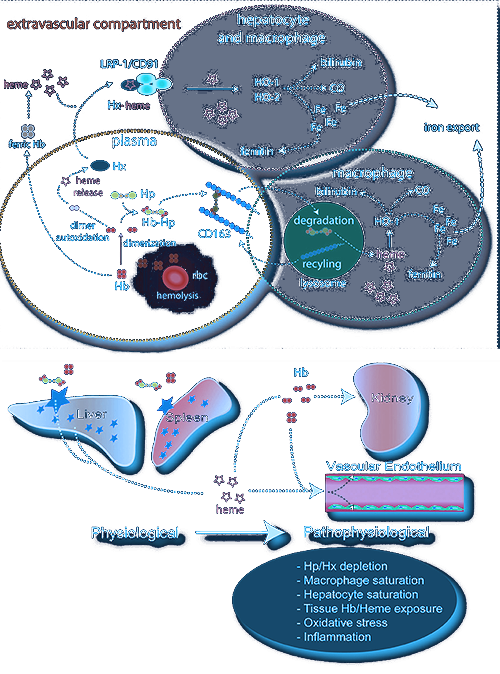
It will be useful to discuss in more detail the dynamics for haptoglobin, given the meaningful role it plays in the containment of cell-free hemoglobin. As stated earlier, haptoglobin binds cell-free hemoglobin, a reaction that cannot happen unless it is in the oxyhemoglobin form. But before you would jump to the conclusion that reducing methemoglobin to oxyhemoglobin would be a desirable outcome, I want to reiterate that the body loses control over haptoglobin production as hemolysis progresses, and oxyhemoglobin in the absence of adequate haptoglobin will most decidedly scavenge nitric oxide and exacerbate hypertension and vascular permeability. There is a very fine line we are walking here with regard to therapeutic strategies. It is vital for a clinician to know what side of that line the patient is on.
As a matter of fact, this is precisely the scenario that we see in severe malaria and one of the ways in which the antimalarial drug chloroquine has proven effective for this disease is via its interactions with hemoglobin.197198 Low haptoglobin levels have been consistently observed in the serum of malaria patients during proteomics analyses.199
Two alleles for haptoglobin production have been identified, Hp1 and Hp2. Hp2 is thought to confer protection against malaria given its correlations with higher haptoglobin levels in less severe or non-symptomatic cases.200 Whether or not this has any significance for COVID-19 remains to be seen. In my opinion, Hp2 alleles would not be expected to protect against severe hemolysis or the resulting cell-free hemoglobin and heme accumulation.
Overloaded Transferrin
In reality, there is a substantial difference between COVID-19 and malaria pathologies. This difference becomes more apparent when we consider transferrin, which binds iron that has separated from hemoglobin, in order to protect against oxidative damage from free iron. While iron-free transferrin is proven to be an antioxidant, protecting lung surfactant against oxidative inactivation, ferric iron-bound transferrin (Fe3+-TF) is known to induce significant inhibition of surfactant, exacerbating respiratory failure.
Increased oxidation of epithelial lining fluid (ELF) amplifies its iron-binding capacity.201202 ELF in normal lungs contains a diverse array of protecting compounds including vitamin E, vitamin C, ceruloplasmin, and transferrin.203 During hemolysis with increased cell-free heme and ferric iron, however, the Fe3+ binds to transferrin in ELF, decreasing surface activity. This further exacerbates vulnerability to oxidative stress and intensifies viral infiltration into AEC’s.
Further, in the presence of hydrogen peroxide, produced as a byproduct of the dismutation of superoxide (which is abundant in oxidatively challenged lung tissue), Fe3+-containing transferrin effectively inactivates the surfactant complex altogether.204 This is an important consideration, because free iron is not exclusive to the domain of hemolysis and breakdown of cell-free methemoglobin — it is also the direct result of AEC injury. Such acute injury can happen with or without hemolysis, during which iron is spontaneously released from ferritin, hemosiderin, and many other such iron-containing proteins.205206207
Hemosiderin, in particular, is abundant in alveolar macrophages, especially in the context of hemolysis, where AMs are engulfing cell-free hemoglobin and degrading it via heme oxygenase, resulting in free iron and biliverdin. The free iron, in turn, is trapped and stored in hemosiderin. The greater the level of cell-free hemoglobin, moreover, the higher the potential for hemosiderin accumulation in the lungs, liver, spleen, kidneys, lymph nodes, and bone marrow — an outcome frequently seen in hemochromatosis.208
On the other hand, iron that has been disassociated from heme will be captured by transferrin, a protein that is up-regulated in the presence of excess serum iron. For this reason, large quantities of it are seen in bronchoalveolar lavage of ARDS patients.209 With increased fluid buildup in the lungs, transferrin will accumulate in both lung interstitium and epithelial lining fluid, where it will bind to ferric iron. In this way, accumulating transferrin “attracts” free iron, creating Fe3+-TF complexes that further degrade surfactant integrity.
In fact, even free ferrous iron will react with hydrogen peroxide in the ELF, creating short-lived, but highly damaging hydroxyl radicals. Such radicals, along with neutrophILderived elastase and phospholipase, will further cleave surfactant components, decreasing activity.210211212
Type II pneumocytes have an abundance of oxidant-scavenging enzymes, including, as I have described above, superoxide dismutase. However, with increased damage to epithelial cells in the airways and decreased surfactant activity, there will be an abundance of antioxidant reactants such as hydrogen peroxide, further elevating the potential for hydroxyl radical formation. This is especially pertinent in asthmatics, who are known to produce excess hydrogen peroxide, detectable in exhalation. In case it isn’t abundantly clear at this point, ferrous iron-containing oxyhemoglobin is capable of reacting with hydrogen peroxide as well and will produce this dangerous reaction in the extravascular space of the lungs, a problem that would actually be exacerbated by reducing agents in the absence of sufficient haptoglobin.213214
Interestingly, serum transferrin of ARDS patients is surprisingly low in iron content, suggesting that the majority of iron bound to transferrin in the lungs may very well be the result of vascular permeability and hemolysis.215 Therefore, administration of recombinant transferrin might also be a valid approach for scavenging iron ions and preventing their destruction of pneumocytes and surfactant. It also stands to reason that individuals with genetic polymorphisms in transferrin expression may be more vulnerable to iron-mediated lung damage, especially in COVID-19 ARDS.
 The “Hemoglobin Attack” Myth
The “Hemoglobin Attack” Myth
Now, I would like to address an unfounded hypothesis that was proffered in an article by a researcher at the Sichuan University of Science and Engineering in China. This article has gotten a lot of attention among pseudo-scientists and hobbyist researchers, and unfortunately, it is now being cited across the internet and on social media as a reflection of fact rather than theory. The article I am referring to uses marginally accurate homology modeling and molecular docking platforms to demonstrate, allegedly, how SARS-CoV-19 attacks cell-free hemoglobin, thereby capturing free heme and inhibiting human heme metabolism. Specifically, they used ExPASy’s SWISS-MODEL and Dassault Systèmes Discovery Studio, both of which are resources more for “experimental exploration” rather than serious, clinically applicable contexts.216
There are several problems with this proposal, but the most glaring problem is the mechanism by which SARS-CoV-2 attack on hemoglobin progresses. First, take a look at the image further above above, which shows the structure of a COVID-19 virion. As should be amply clear at this point, viral RNA is transcribed into proteins only after it has entered a cell, having gained access to the ribosomal machinery. Prior to cell entry, it is enclosed in nucleocapsids inside the structural envelope. As such, the open reading frame (ORF) RNA segments are not viral “proteins” until they have been translated by host cell ribosomes. This is a critical consideration when reading the Sichuan University proposal, as the entire basis for hemoglobin attack is based on viral ORF “proteins”.
Now, consider that an intact virion, prior to its infiltration into host cells, exposes only its S, E, and M proteins, with the S “spikes” being the most prominent. Together, they comprise the “envelope”. To be clear — there are no ORF proteins presented on the surface of the virion, because, quite simply, they haven’t yet become proteins! They are still “dormant” in the virion RNA contained within the nucleocapsid (N). Further, as I will explain below, these ORFs have not been observed in extracellular complexes nor in a viable state outside of infected cells. Moreover, no proof exists whatsoever that coronavirus virions possess the capability to enter red blood cells, where hemoglobin resides in the majority of cases — nor has any compelling evidence surfaced that would indicate excessive hemolysis and accumulation of cell-free hemoglobin. The glaring question is, how could the ORF proteins be exposed, in the first place, to hemoglobin? Even if they were able to enter red blood cells (where RAS system membrane receptors have been described), mature RBCs do not contain a nucleus, ribosomes, or mitochondria. There is absolutely no machinery present to translate the viral RNA into functional proteins. Hopefully, you are beginning to see the glaring error in this paper’s premise.
Next, consider the detailed hypothesis: ORF8 along with the small (E2) surface glycoprotein binds to cell-free hemoglobin. At the same time, ORF1ab, ORF10, and ORF3a “proteins” coordinate an attack on the 1-beta chain of hemoglobin to dissociate iron and release free heme. This attack purportedly causes “less and less hemoglobin that can carry oxygen and carbon dioxide”.
For a non-scientist or average reader with limited knowledge of heme dynamics and/or virology, this hypothesis might seem quite disturbing. Unfortunately, in order for this to work, we have to imagine, somehow, all of these ORF proteins in a cluster or complex that gains access to cell-free hemoglobin. To be quite blunt — this has never been demonstrated either in vitro or in vivo. Further, there is no possibility for virions to expose these proteins to hemoglobin inside red blood cells due to the lack of ribosomal machinery. Truly, this theory is wildly hypothetical and could only have been imagined using “in silico” models — i.e. computer simulation. Indeed, the author of this paper, analyzed the ORF proteins in isolation, observing their binding patterns with hemoglobin, free-heme, and other human molecules that have known interactions with them.
Further, if you will read this paper in its entirety, you’ll find that the author modeled the interactions of the spike and E proteins with heme first, and they were shown to have no affinity. Right away, this should raise a red flag for the astute reader. The author continued the investigation by analyzing nucleocapsid binding with free-heme. At this point, we are firmly in the realm of fantasy, as there is no possible way viral nucleocapsids would have contact with cell-free heme outside of their envelopes. Were this the result of viral envelope disruption, resulting free-floating, dismembered proteins would be promptly phagocytosed, extracellularly.
Nonetheless, this did not deter the author from modeling viral proteins ORF1ab, ORF3a, ORF6, ORF7a, ORF8, and ORF10 with human heme-binding proteins. In fact, it was determined that they all, with the exception of ORF6, have conserved functional domains with these human proteins. Of note — the domains in the five viral proteins were different, suggesting they might not bind heme in “exactly the same way”.
To make matters worse, ORF1ab, ORF10, and ORF3a demonstrated a complete inability to bind with heme in the author’s model until iron was removed. This led to the rather illogical presumption that instead of binding, they collectively attack hemoglobin and then “pry and kick” iron out of it. Once iron has been freed from hemoglobin, the author proposes (also based on nothing more than binding affinities), that ORF6 and 7 “pass” the free heme along to ORF8, which hugs it tightly and whisks it away “for use by the virions.” This is on par with an animal’s intestines running off to hunt down prey. In a stroke of pure poetry, this paper summarizes the process as follows: “the novel coronavirus targeted hemoglobin, attacked heme and hunted porphyrins”. Mind you — all in silico on a university computer with zero peer reviews or basic follow-up with in vitro experimentation.
In the first draft of the paper, this pseudo-scientific story was rounded out with a description regarding why chloroquine is desirable for resolution of this problem, as — according to further modeling — it shows the capability of competing with free-heme to bind with the viral protein, thereby inhibiting the viral protein’s attack on heme by binding to it. Not surprisingly, this assumption was rescinded after the author endured multiple attacks from more astute readers in the scientific community (see the footnotes in the article).
To reiterate, the entire premise of this paper is based on the ability of viral proteins — generated by host ribosomes after cellular invasion — to interact with cell-free hemoglobin outside the same cell. There are few indications, to date, that such proteins have significant actions outside the infected cell’s interior. For example, though SARS-CoV ORF3a protein has been detected in lung specimens of infected patients along with corresponding antibodies, this is not evidence of significant extracellular activity. On the contrary, ORF3a is primarily localized in intracellular / plasma membranes and, along with other ORF proteins, activate inflammasomes, affect nuclear transcription, and — in the case of B lymphocytes — drive antibody production. That is, there is a predominance of evidence supporting ORF protein modulation of host immunity from within its cells, rather than outside.217218
Nonetheless, ORF3a has been proven to bind to the spike protein, intracellularly, in the process of being incorporated into virus-like-particles — along with M and E structural proteins. Further, infected cells have also demonstrated the release of membrane-bound ORF3a structures, similar to sole E protein structures — however, what is being proposed in this paper is a coordinated attack on hemoglobin by multiple ORF proteins, in concert, outside of an infected cell. If this attack were to be happening directly inside non-hemolysed red blood cells, then we have to clear the hurdle of explaining how this is even functionally possible since 1) red blood cells contain neither a nucleus nor ribosomes and so cannot translate viral RNA into functional proteins and or respond to transcriptional cues and 2) no such infection of RBCs has been observed across the hundreds of studies to date published on this disease. Further, though this full array of viral proteins would be expected in bronchoalveolar lavage fluid, partially due to pyroptosis, apoptosis, or even viral envelope disintegration, the idea that they are chained together — either as a macromolecular membrane-bound structure or free-floating, “traveling in packs” like wolves — is, quite frankly, bordering on insanity.219220221222
There are countless other details in this paper that demonstrate a grave paucity of knowledge in even basic molecular biology, including lack of acknowledgment of an alternate high-affinity ACE2 binding site, the prevalence of porphyrins in urine/saliva/feces (as opposed to being broken down into biliverdin), increased host cell infection for porphyrin / heme-bound virions — and many other patently false, grossly ignorant assertions.
As it turns out, the author eventually uncovered a modicum of conscience and amended the original paper with some revealing commentaries. In particular:
“This paper is only for academic discussion, the correctness of the theory needs to be confirmed by other experiments. According to the reader’s suggestion, the content of the drug-related efficacy analysis has been deleted. Due to the side effects of drugs, please consult a qualified doctor for detailed treatment information, and do not take the drug yourself. We look forward to these discoveries bringing more ideas to people and inspiring people’s confidence in defeating the virus.”
In other words — this is science-fiction, yet the author still hopes there will be scientists foolish enough to test the theory out in a petri dish. Whether or not that will happen remains to be seen.
My only reason for bringing attention to this paper is because it is a perfect example of how unproven ideas, stated in complex scientific language, can misguide and confuse readers, especially those with a lack of thorough grounding in molecular/biological science. For this reason, it is my suggestion that you sidestep the opinions of bloggers, “biomedical researchers”, and other writers that in any way refer to this particular paper in a manner that might convey it as fact. Citing experimental research in such a context demonstrates a lack of ability to properly discriminate experimental models from statistically significant lab / clinical results, thereby invalidating anything else that particular source might have to say, at least as far as COVID-19 is concerned.
Heme / Hemoglobin Confusion
In this vein, I would also like to propose that you pay very close attention to the terminology your information sources use when describing the COVID-19 pathology, particularly with respect to cell-free hemoglobin. I have noticed consistent confusion in language surfacing in blogs and social media outlets that describe redox states of iron and its association with heme. To summarize and help clarify what we’ve been discussing thus far, the following are the stages of iron dissociation from hemoglobin:
- Hemolysis releases hemoglobin from red blood cells either bound to oxygen (oxyhemoglobin) or without oxygen (deoxyhemoglobin), both of which contain a ferrous iron “core” (Fe2+). It is, at that point, “cell-free”, though it will be far more “reactive” in the oxyhemoglobin form.
- Once free, oxyhemoglobin is either bound by haptoglobin and taken up by macrophages for degradation or proceeds to deleterious reactions.
- Oxyhemoglobin reacts with either nitric oxide to form nitrate, or with hydrogen peroxide in a Fenton reaction to form a hydroxyl radical. In doing so, it becomes methemoglobin (with an oxidized ferric iron core (Fe3+)).
- Methemoglobin can then oxidize to ferryl hemoglobin (Fe4+) and cause damage to lipids or completely dissociate from its ferric Fe3+ iron ion and become free heme, leaving the ferric iron to further react or be picked up by transferrin.
- Free heme can inflict further oxidative damage if it is not scavenged by hemopexin or degraded by heme oxygenase into biliverdin and passed to the liver for excretion.
- Free ferric iron can induce oxidative damage and activate immune responses if it is not taken up by transferrin, though if bound to transferrin in the lung, it will reduce surfactant and increase potential for further lung damage.
Pay close attention to those writers that interchange “heme” and “hemoglobin” in the context of describing this process. They are not the same. To understand the context for any given therapeutic strategy, these hemodynamics must be understood without any confusion, whatsoever. Otherwise, the wrong steps may be taken at the wrong time, and it could have dire consequences, as we will discuss in further detail below.
Elevated Coagulation
Another mechanism that can complicate hemodynamics in COVID-19 is a decline in antithrombin and increased platelet aggregation/coagulation.223 This, in part, is an antimicrobial defense mechanism that is over 400 million years old, back when insects were using Toll-receptor signaling to detect invaders and release antimicrobial peptides during hemolymph clotting.224225226227228
In COVID-19, it can become so widespread — again, depending on viral load — to the point of disseminated intravascular coagulation, which can increase mortality substantially.229 Therefore, considering antithrombin levels and perhaps also looking at prothrombin / prothrombin time (PTT) would provide insight into the risk for not only thrombosis, but also complications associated with increased vasoconstriction as a result of ACE2 shedding and increased AngII.
Stage IV: Hypoxia and Acute Respiratory Distress
Hypoxia-Inducible Factors: A Balancing Act
Another important system which plays into the severity of ARDS is the HIF-system or “hypoxia-inducible factors”. This is a collection of transcription regulators in cells that respond in the absence of sufficient levels of oxygen (i.e. hypoxia). The primary gene responsible for orchestrating these transcription regulators is HIF-1alpha, a subunit of the HIF-1 complex. Optimal expression of HIF-1a is absolutely mandatory for the maintenance of oxygen delivery to tissues and organs during acute respiratory distress. In the majority of cases, even optimal expression is inadequate to address the hypoxia resulting from widespread type I pneumocyte destruction and lowered gas exchange in alveoli.
Though detrimental effects can be seen from HIF-1a over-expression, this is generally only a problem in the context of cancer or tumor metastasis, which hijacks HIF-1 expression to increase oxygen supply to such cellular colonies.230231 In all other contexts, especially ARDS, down-regulation of this system, by any means, must be avoided. Such HIF-1 inhibitors include nitric oxide deficiency, lack of oxyhemoglobin, and ascorbate excess, all of which can have deleterious effects on this system during the progression of this disease.
Once HIF-1a has been activated, it invokes an up-regulation in nuclear transcription for a number of genes that increase oxygen delivery, including erythropoietin for RBC production, transferrin for control of free iron in plasma, VEGF — which increases vascular capacity via angiogenesis, heme oxygenase for degradation of free heme, and both inducible and endothelial NOS for increased nitric oxide production and improved vascular tone.232
Throughout the hypoxia response, prolyl hydroxylase domain-containing protein 2 (PHD2), which serves as the predominant oxygen sensor in cells, constantly informs other cells of oxygen status. It does this in part via its high binding affinity for ferrous iron and alpha-ketoglutarate (generated primarily from the Kreb’s cycle). As both Fe2+ and AKG levels rise, PHD2 binds with them, altering its configuration to allow hydroxylation of HIF-1a at two proline residues. This HIF-1a hydroxylation allows VHL (von Hippel-Lindau protein) to recognize and bury it in its hydrophobic core, where it can be subsequently degraded. This, in turn, “turns off” transcription for the anti-hypoxia factors.
It was recently demonstrated that L-ascorbate (i.e. vitamin C) is capable of competing with alpha-ketoglutarate for the binding site on PHD2, thereby inducing, along with ferrous iron, the hydroxylation of HIF-1a. As a matter of fact, this behavior has been leveraged in studies attempting to mitigate tumor metastasis — by increasing VHL degradation of HIF-1a and shutting down hypoxia-inducible factors. We’ll discuss this problem in more detail below in the section on natural therapeutics, but suffice it to say now that high-dose ascorbate, especially intravenous administrations, could very well be expected to blunt hypoxia response and exacerbate respiratory distress, both via directly inducing HIF-1a hydroxylation by binding to PHD2 as well as reducing ferric iron to ferrous form, thereby completing the “circle of deactivation” for this enzyme.
As has already been mentioned earlier, airway epithelial cells have the capability of producing hepatocyte growth factor (HGF), which also enhances HIF-1a expression at the transcriptional level.233234 Another aspect of HIF-signaling that is important to understand is the way in which nuclear respiratory factor 1 (NRF-1) inhibits HIF-1a translation. NRF-1 is another transcription factor that activates the expression of nuclear genes required for respiration, heme biosynthesis, and mitochondrial DNA transcription and replication.235 When there is excessive oxidative stress, we see a resolute stabilization of PGC1a, a master regulator of mitochondrial biogenesis. PGC1a stabilization, in turn, activates NRF-1.236
In this way, as oxidative stress provokes HIF1-a transcription, it also stabilizes PGC1a and activates NRF-1 to provide additional “dampening” effects on excessive HIF-1 induction — in other words, it is a negative biofeedback regulator. In fact, there are binding sites specifically for NRF-1 on the HIF1-a protein, so it appears to have an even more direct inhibitory function than PHD2 / VHL-mediated degradation (which is primarily driven by oxygen-sensing).
It is important to note that HIF1-a is activated not only by oxidative stress, but angiotensin II as well. Therefore, as COVID-19 provokes ACE2 shedding and angiotensin II levels rise, there is increased potential for HIF-1a activation— and for very good reason. Without such activation, the body would be starved of oxygen!237238239 Nonetheless, individual genetics for mitochondrial biogenesis, as well as NRF-1 expression, must be considered when quantifying the risk for HIF1-a over-expression. In my opinion, ARDS presents a far greater risk for inadequate hypoxia response, regardless of mitochondrial dynamics.240241
This brings me to the discussion of another myth that has arisen, apparently through the same channels that have been promoting SARS-CoV-2 “hemoglobin attack” theories. The myth I am referring to relates to recent evidence that the SARS-CoV-2 spike protein contains a furin-like cleavage site. It has been proposed that this could be a problem by virtue of the fact that multiple hypoxia-inducible factors are activated by furin cleavage, and the up-regulation in furin expression could potentially “increase cleavage of CoV-2 spike proteins”, thereby amplifying virulence. There are a number of things wrong with this hypothesis, so let’s break it down.
Under hypoxic conditions, HIF-1 has been demonstrated to up-regulate the expression of furin via its promoter region. Furin is known to have multiple substrates, including cytokines, hormones, growth factors, and a wide array of receptors. Its activity is notably up-regulated in diseases such as cancer. In the case of hypoxia, furin is primarily seen to be activating cadherins/integrins, and TGF-beta, but its expression may also be directly modulated by inflammatory processes. In other words — regardless of hypoxic status, in an inflammatory environment, we are going to see elevated furin activity. It’s simply unavoidable, especially in inflammatory crises such as COVID-19 ARDS.242
The problem with furin-family enzymes is that they are veritable “Swiss army knives” that perform literally hundreds of discrete functions. The more furin functions are studied, the more it is made obvious that they can’t be avoided, especially in viral infection. It should come as no surprise that furins not only activate hormones, growth factors, and enzymes, but they are also used for the proteolytic activation of viruses and bacteria. In particular, coronavirus spike proteins are cleaved and “primed” for cell attachment by furins as well.243 One of the ways that cells deal with this problem is by up-regulating interferon response to inhibit the enzymatic activity of furin-like enzymes.244
That being said, furin proteins are highly expressed in the lungs, and viruses that infect the respiratory tract such as SARS-CoV-2 are able to leverage furin to activate surface glycoproteins. The higher levels of inflammation and pneumocyte damage rise, the more we are going to see furin-mediated HIF-1a up-regulation. Therein lies the basis of the myth circulating around the internet that “HIF1 promotes furins, furins cleave CoV-2 spike proteins, so it should be desirable to inhibit HIF-1.”245246 This may sound good in theory, until you consider the implications of broad HIF-1 inhibition. As I’ve mentioned above, such inhibition is desirable only in clinical contexts in which there is a known cancer or tumor metastasis. The entire correlation between HIF-1 and virally-leveraged furin cleavage is extraordinarily weak, based only on the coincidence of furin expression in each scenario.
The truth of the matter is that you’re going to see furin up-regulation in any hyper-inflammatory state (e.g. cytokine storm). It cannot be avoided, and the fact that viruses and bacteria leverage its expression to gain advantages is a purely secondary concern. HIF-1a activation is a normal and desirable outcome in the ARDS milieu, without which there would be severe oxygen deprivation. The body has developed ways of ensuring balance in this system, not the least of which is NRF1-mediated negative regulation. Suppressing HIF-1 invocation for the purposes of avoiding furin-mediated spike protein cleavage (and higher viral replication rates) is on par with provoking HIF-1 using hyperbaric oxygen therapy because cancer cells “may only thrive in a low-oxygen environment”. The former ignores HIF-1’s role in reversing oxygen deprivation, while the latter clearly misunderstands HIF-1’s relationship to tumors.
Therefore, similar to the “hemoglobin attack” myth mentioned earlier, the idea that hypoxia-inducible factors are undesirable in a full-blown ARDS pathology due to increased furin activity should also be quickly dispensed to the category of “dangerous pseudoscience”. The way forward way is not to inhibit the body’s response to oxygen deprivation, if and when that occurs, but rather to target other pathways — for example interferon expression and PGC1a / NRF1 enhancement — that can reliably confer greater resilience against viral infection and keep HIF-1 signaling under control.
Pulmonary Fibrosis and Hypertension
Clearly, when considering therapeutics for COVID-19, we should be less concerned about homeostatic mechanisms the body uses to maintain balance, such as HIF-1, and focus more on the original problem: compromised immunity, loss of inflammatory control, and resulting pulmonary fibrosis. These are the provocateurs of hypoxia, and the sooner we address these concerns directly, the less we will have to worry about secondary issues such as derangement in furin expression.247
One of the biggest problems that occurs at this stage is the increasing mass of alveolar epithelial cells and extracellular matrix deposition within the airspace. As mentioned earlier, this damages both type I and II AECs along with decreased protection of alveolar collapse by surfactant.248 In this scenario, both epithelial and endothelial cells are throwing oil in the fire, especially during viral infection where there is poor antiviral control. To reiterate, coronaviruses exacerbate inflammation via their activation of inflammasomes. This is a non-HIF correlated process, and the majority of furins we see in COVID-19 are the result of NF-kappaB induction and resulting promotion their expression.249
In addition to inflammatory cascades, complement and coagulation pathways are also activated, and this provokes lung tissue to release even higher amounts of reactive oxygen and nitrogen species. This is how the so-called “cytokine storm” escalates.250 So at this stage, we should be less concerned with the signaling cascades induced by excessive inflammation, and concentrating more intently on proper strategies to prevent this inflammatory runaway train in the first place.251252253254255
Even during the early stages of COVID-19, alveolar macrophages provide an early response to danger signals from AECs via their production of chemokines and cytokines. This, in turn, recruits polymorphonuclear leukocytes (i.e. neutrophils).256 In particular, elevated levels of CCL2 have been observed in bronchoalveolar lavage fluid. This chemokine is known to recruit monocytes, T-cells, dendritic cells, and fibrocytes to areas of inflammation. The fibroblasts differentiate into myofibroblasts and extracellular matrix proteins — mostly collagens — followed by their secretion into the alveolar space, where they accumulate in the parenchyma, leading to fibrosis. Therefore, the ideal window to prevent ARDS is during the initial acute inflammatory response, mediated by macrophages.
If we can adopt strategies that will prevent the escalation of pro-inflammatory processes early on in the course of this disease, we can avoid the devastating changes that happen to the lungs as a result of the fibroproliferative phase. To further clarify where we are in the process in this discussion, consider that significant furin up-regulation (i.e. enough to substantially increase spike protein cleavage and viral infection) is not seen until either severe hypoxia or significant cytokine/chemokine activity. Therefore, it is safe to say that the most optimal therapeutic window is prior to HIF-1 induction.
Nonetheless, we have here the perfect example of how HIF-1 signaling can be protective. One of the transcription factors up-regulated by HIF-1 is angiopoietin-1 (ANGPT1) along with its endothelial-specific receptor tyrosine kinase TEK). Together, these factors increase angiogenesis and development of fresh, new vasculature.257 ANGPT1 has shown the ability to prevent endothelial permeability and apoptosis, thereby improving the integrity of the vasculature, and it does this without inducing growth responses in endothelial cells.258259260
Similar protective effects have been seen with keratinocyte growth factor, administered in models of idiopathic pneumonia. In such contexts, KGF suppresses T-cell-mediated macrophage activation and pro-inflammatory cytokine release.261 As such, it may have therapeutic potential. The challenge with such therapies would be delivering KGFs effectively to the lung. This problem was addressed in a 2008 study at the London Research Institute that demonstrated, for the first time, how mesenchymal KGF-expressing stem cells could be used to effectively reverse chronic obstructive pulmonary disease (COPD).262 A few years later, CCL2-inhibited mesenchymal stem cells (MSCs) were delivered intravenously to mice with pulmonary fibrosis, showing their ability to significantly reduce neutrophil infiltration in the lung and reduce plasma IL6 and IL1B, two cytokines frequently seen in the most severe cases of COVID-19 ARDS.263
Therefore, it could be that recombinant angiopoietin in combination with CCL2-inhibited and/or KGF-enhanced mesenchymal stem cells could powerfully block neutrophil infiltration, vascular permeability and capillary leakage, and lung edema, all of which, incidentally, are associated with down-regulated ANGPT-1, a primary transcription factor of HIF-1 signaling. Knowing this, do we still believe that inhibiting HIF-1 signaling to lower furin expression is a viable option to control viral replication?264 Other scientists have been thinking along the same lines for quite some time now, as demonstrated in another 2007 study showing a skin fibroblast-based ANGPT-1 delivery system in models of acute lung injury. Not surprisingly, a marked improvement in lung inflammation injury was noted.265
Another plus to using MSCs for the treatment (or prevention) of SARS-CoV-2 associated lung injury is that they are innately “hypo”-immunogenic. That is, because they express low levels of major histocompatibility complex (MHC) class I and II and completely lack expression of co-stimulatory molecules, they would not be expected to produce an immune-mediated inflammatory response. MSCs, further, may be isolated from bone marrow, adipose tissue, umbilical cords, and even lung tissue with a very high yield — moreover, they can be easily multiplied in vitro.
What makes them a particularly attractive option for the treatment of acute lung injury, especially as provoked by COVID-19, would be their capability of being genetically modified to “home in” on injured sites. To date, there have been significant challenges in achieving adequate engraftment rates to realize therapeutic benefits.266267268
Nonetheless, the applications are promising, as MSCs could be modified in an endless number of ways to achieve desired outcomes. For example, heme-oxygenase modified MSCs (MSCs-HO-1), have proven antioxidant, anti-inflammatory, and antiapoptotic effects in models of acute lung injury. Consider that in later states of COVID-19 ARDS, there may be a higher occurrence of hemolysis and parallel cell-free heme, the latter of which could be resolved with AEC-targeted HO-1 expressing MSCs.269
At this stage, the most detrimental change has already occurred in the vasculature as a result of deranged nitric oxide signaling. It is specifically the increase in blood pressure as a result of vasoconstriction that leads to vascular permeability in the first place, pushing blood cells into the extravascular space, often inducing hemolysis and the pro-inflammatory effects of cell-free hemoglobin described above.
Therefore, it could be said that the single most detrimental development in the course of this disease is the depletion of nitric oxide which can, in part, by exacerbated by the presence of cell-free oxyhemoglobin — that is, oxygenated ferrous “oxyhemoglobin”. It is specifically this form of hemoglobin that scavenges nitric oxide and provokes vasoconstriction and dangerous spikes in blood pressure. Such oxygenated hemoglobin is ultimately only useful when encapsulated in red blood cells. This does not mean I am underestimating the deleterious effects of oxidized cell-free hemoglobin (methemoglobin). I just want to be very careful to underscore the importance of context when interpreting other discussions on COVID-19 and its potential influence on hemodynamics.
I have reiterated this point many times, but it bears repeating: cell-free oxyhemoglobin has not been proven to be the primary progenitor for COVID-19 ARDS, and many other mechanisms have been fully elaborated to explain disease progression. Nonetheless, we cannot ignore the heightened potential for vascular permeability at later stages of the disease, following by increased scavenging of nitric oxide. This results in increased methemoglobin, especially in the absence of adequate haptoglobin response. Further, oxyhemoglobin does not deliver oxygen in the same manner as its RBC-encapsulated counterpart. In fact, oxyhemoglobin delivers oxygen prematurely to arterioles, a mechanism which makes it 1,000 more effective at scavenging NO.270
To make matters worse, cell-free oxyhemoglobin extravasates (i.e. leaks) through the endothelial layer into tissues (e,g, alveoli in the lung) where it further directly scavenges NO and/or is catabolized to heme and globin, the latter of which is highly inflammatory.271272 Stated simply, cell-free oxyhemoglobin induces both systemic and pulmonary hypertension and is potentially far more detrimental than methemoglobin, as a whole, to the ARDS patient. As previously mentioned, it is specifically for this reason that cell-free hemoglobin-based blood substitutes (e.g. HBOCs) are correlated with hypertension.273274275276277
The higher the cell-free hemoglobin volume, the greater the possibility there will be for haptoglobin and hemopexin depletion. If reducing agents (i.e. antioxidants) are administered in this milieu, there will be a severe exacerbation of NO scavenging, oxyhemoglobin extravasation, and increased oxidative stress in extravascular tissues, particularly alveoli.278279 In fact, high levels of circulating cell-free methemoglobin do not generally affect hemodynamics if a patient’s urate, ascorbate, or glutathione status are compromised. It only becomes vasoactive in the presence of these antioxidants, due to the conversion back to NO-reactive oxyhemoglobin. Further, methemoglobin inflicts its greatest damage in the extravascular space, after oxyhemoglobin has increased vascular permeability.280281282
In other words, urate, ascorbic acid, and glutathione in excess would all be expected to worsen ARDS outcomes, due to their oxyhemoglobin enhancing effects and resulting impairment of endothelial function.283284285 For this reason, in order to avoid vascular injury, increased permeability, and further oxidative damage in the extravascular space (i.e. alveoli), it is important to avoid aggressive administration of reducing agents — especially in combination with nitric oxide therapy — thereby lessening the burden on haptoglobin, and reducing vasoconstriction.286 The resulting excess methemoglobin will be cleared faster due to its lower stability, catalyzed to heme and ferric iron which, consequently will be picked up by splenic macrophages and transferrin, respectively.
Further clarification here is needed, since there have been numerous recommendations lately for the use of reducing agents, such as ascorbic acid, for the reversal of COVID-19 ARDS. Please, follow due diligence and do your best to understand hemodynamics as best as you can. Oxyhemoglobin is not going to carry oxygen to cells. It is NO-reactive outside the RBC and will only oxygenate tissues when properly encapsulated. As I have mentioned earlier, RBCs contain antioxidants that stabilize oxyhemoglobin intracellularly — this is not so after hemolysis.
Therefore, reducing methemoglobin with, for example, ascorbic acid will actually deprive the body of oxygen by scavenging NO, increasing oxyhemoglobin extravasation into alveoli and then — oxidizing to methemoglobin extravascularly. The important nuance to understand here is that methemoglobin is not as big of a problem intravascularly as outside the vasculature. It is primarily reduced cell-free oxyhemoglobin that causes vasoconstriction, vascular permeability, and cell-damaging effects in alveoli. Haptoglobin-mediated clearance is far more efficient in the intravascular space. Oxidizing reactions in the more crowded, hyper-inflammatory milieu of airway epithelial cells, alveolar macrophages, and the various other cascades I’ve described above are considerably more damaging than if vascular permeability was avoided in the first place.287288
If reducing agents are to be used, care must be taken to determine the stage of disease progression. Whereas they could afford some anti-inflammatory aid early on by containing oxidative processes, once angiotensin II levels have reached a certain ceiling and frank hypertension has set in, the risk for vascular permeability increases, along with hemolysis, contraindicating the use of anti-oxidants for the reasons stated above. It can only be presumed that the improvements seen in sepsis-models with high-dose reducing agents are attributable to the lack or complete absence of hemolysis and cell-free oxyhemoglobin.
Moreover, the higher a patient’s antioxidant status, the faster the conversion from methemoglobin to oxyhemoglobin will be. One study demonstrated this unequivocally by separately infusing both metHb and oxyHb in serum. On a micromolar basis, metHb was shown to form oxyHb at a significantly faster rate than oxyHb to metHb — and this effect is seen without antioxidant supplementation. This effect was, in part, presumed to be the result of metHb dissociating faster, favoring the formation of dimers with heme, which in turn release heme for degradation to biliverdin.
Further, a higher percentage of dimers present might also increase clearance by haptoglobin.289290 With oxyHb, on the other hand, it will scavenge all available NO in a matter of milliseconds, far faster than haptoglobin would have a chance to bind and sequester it. Reducing agents would further “stabilize” hemoglobin in the oxyHb state, inducing excess reactivity with NO.
COVID-19 Medical Interventions: Pros and Cons
 Now that we’ve established the broad stages through which COVID-19 progresses, let’s do a deep dive into therapeutic strategies, both pharmaceutical and natural/alternative approaches, to better understand their pros and cons in a clinical context
Now that we’ve established the broad stages through which COVID-19 progresses, let’s do a deep dive into therapeutic strategies, both pharmaceutical and natural/alternative approaches, to better understand their pros and cons in a clinical context
The majority of proposed therapies to date have centered around inhibition of viral entry into cells. This has resulted in strategies that include blocking the ACE2 receptor and/or the viral spike-protein directly. We have already discussed the challenges of vaccines, particularly the potential for antibody-dependent enhancement of viral infection in certain vulnerable subsets of people.
There are, however, other immune-modulating therapies currently being discussed including polyclonal antibodies via plasma therapy, polypeptide hormones for the maturation of T-cells, development of ACE2 “immunoadhesins”, and even IL6 targeted monoclonal antibodies.
Clinics around the globe have been busy designing and developing advanced protein production platforms using advanced expression analytics. The main goal is to provide efficient monoclonal antibodies at an affordable cost without long delays.291 Despite progress in the development of monoclonal antibody-based passive immunotherapy, no monoclonal antibodies are yet available on the market. This is, in part, due to the labor-intensive, expensive, and time-consuming process required for their production.
While viral vectors, nanoparticles, activated whole virus / DNA vaccines, and monoclonal antibodies have all been used effectively for SARS-CoV, we have a new playing field with SARS-CoV-2 possessing unique genomic aspects. For this reason, what has worked in the past may not be a silver bullet for our present challenges and, more than likely, will need to be extensively adapted, given the nuances in disease progression that I described in this article.292
At this time, the development of vaccines and other therapies continues, but we nonetheless need to focus on other multi-pronged strategies to deal with symptoms and prevent further deaths. And while the death counts do seem to be declining at the time of this article’s release, virologists are nonetheless warning of the high rate of viral genomic mutation that is occurring and the possibility for potentially severe changes in adaptation to human immune defense.293
This underscores one of the other reasons I felt this article was necessary: we need to gather all the facts available to better understand the means by which SARS-CoV-2 has adapted and improved its anti-immune blockade. Given the short amount of time (less than two decades) that these mutations have occurred — and the constantly shifting canvas of technological advancement, with or without assessment of biological effects on human health, there is a high possibility for not only a second wave, but multiple branches with a widening repertoire of adaptations to not only human immunity but that of other species as well
So while antibody defense and “herd immunity” is certainly desirable in the short-term, it nonetheless does not protect us from rapid mutations and resulting adaptations that would quickly and perhaps unexpectedly sidestep our best efforts for protection.
Current Proposals from Around the World
Vaccines
There has been a lot of talk about antibody response, in particular to the spike protein. At the time of this article, over 150 tests have appeared globally, only a fraction of which could be considered reliable and accurate. Nonetheless, there has been a great deal of misunderstanding about the protections that antibodies confer, especially post-infection or post-recovery. Even during the process of infection, anti–spike IgG antibodies may directly induce inflammatory responses.294
This is especially prevalent in the later stages of coronavirus infection, as demonstrated with SARS-CoV, which was shown to cause acute lung injury persisting until the latest stages. This is an extremely important consideration for those that would argue the viability of a SARS-CoV-2 vaccine. This article is not meant to raise the time-weathered debate of vaccine side effects. That being said, it should be noted that the SARS-CoV vaccine directly induced pulmonary injury during testing with mice and African green monkeys, but the medical community ignored these warning signs and pushed forward with vaccine development.295296297
Not surprisingly, similar inflammation-mediated lung injury was noted in human patients that received the vaccine as well. As far back as 2003, it was determined that the development of acute respiratory disease coincided with antiviral IgG seroconversion in as much as 80% of the patients.298 More troubling is the fact that those patients who developed an anti-S-neutralizing antibody faster actually had a much greater risk of dying from SARS-CoV. To put this in perspective, it took about 15 days for the deceased group to reach peak antibody levels, while those that survived developed antibodies in ~20 days. Only a 5-day difference delineated mortality risk. Interpret that as you will.299
One of the explanations for this unexpected response was how the presence of spike-IgG altered alveolar macrophage response prior to viral clearance. It was shown that anti-Spike IgG provoked accumulation of both monocytes and macrophages, leading to excess production of MCP-1 and IL8 in the lungs. It appears that this inflammatory cascade was triggered by the IgG complex binding to Fc immunoglobulin receptors on the monocytes/macrophages — and was confirmed with Fc receptor blockers, which significantly reduced inflammation.
So the question is: why do some patients produce the neutralizing antibody early, leading to chronic inflammation, ARDS, and even death — while others survive with relatively few complications? I believe this could, in part, be explained as an antibody-dependent enhancement (ADE) — a phenomenon in which sub-optimal antibody response enhances viral replication inflammation, prior to viral clearance. ADE has been demonstrated in a wide variety of viral infections ranging from dengue and flavivirus to even influenza.
Following virus-antibody complex interaction with Fc receptors, ADE essentially promotes their intracellular uptake. This, in turn, leads to the enhancement of infection in the target cell.300 For this reason, vaccine-provoked antibody production, especially if administered in the presence of active infection, could have dire consequences.
In healthy individuals with strong antiviral response (i.e. younger populations with more robust hematopoietic stem cell reserve), neutralizing antibodies can further diminish viral replication. However, in an immunocompromised individual or someone who has been unable to effectively suppress viral replication to any degree, premature antibody production could lead to internalization via Fc receptors and further enhancement of viral infection.301
Antibody response to SARS-CoV-2 is still being unraveled, though the consensus to this point is that S1 antigens are more specific to CoV-2 than S, which has proven cross-reactive with MERS-CoV spike protein. For this reason, S1 has been the antigen of choice for SARS-CoV-2 diagnostics.302303
Since it has been approximately 17 years since SARS-CoV was circulating in the human population, there has been the gradual disappearance of SARS-CoV-specific antibodies. This made them virtually undetectable in up to 91% of samples tested 6 years after infection. Therefore, the presumption has been made that false-positives for CoV-2 with SARS-CoV-1 are statistically unlikely.304 The same may not be said, however, regarding cytomegalovirus, Epstein-Barr virus, or Mycoplasma pneumoniae, all of which have shown higher potential for false-positive results.305
Therefore, it became necessary in recent months to step away from spike protein antibodies and focus, instead, on the N (nucleocapsid) protein, which has only 90% similarity with SARS-CoV. Fortunately, N-antibodies are of higher specificity. This does not, however, exclude the potential for cross-reactivity. And to complicate matters, not every antibody test has been third-party tested or approved by regulatory agencies.306307
 Antibodies
Antibodies
A neutralizing antibody targeting the S proteins on the surface of the Cov-2 virus has been proposed as a means of providing passive immunity.308 This may be a faster route than vaccines, because it could use antibody fragment-containing phage or yeast to quickly zero in on potential candidates for viral neutralization.309310 The problem here is that each candidate would need to be exhaustively tested, both in vitro and in vivo (i.e. animals) to confirm potency.
This approach would also lead to the need to use multiple recombinant antibodies in a “slurry”, in order to achieve broader protection in humans. This, in and of itself, raises a red flag, if not for the fact that successful in vitro / animal testing does not inherently confer viability for use in humans. Further, I don’t see that the labs and clinics considering this option are prepared for the comprehensive due diligence required for assessing individual reactions to each antibody, given pre-existing conditions and genetic genotypes.
An alternate, similar strategy has been proposed that would immunize larger animals (e.g. sheep, goats, or cows) and then extract and purify polyclonal antibodies from those animals. The problem with this approach is subtle but nonetheless significant differences in immunological genetics between those animals and humans.311 Further, using either of these recombinant antibody approaches, there will always be the potential for human immune system reactivity to foreign immunoglobulins.312
Therefore, this strategy, in my opinion, is dead in the water before it’s even gotten to the design phase. There is no possibility we can risk widespread, unexpected immunological chaos in human populations at large. No amount of testing in vitro or animals could prevent that possibility. Moreover, it would clearly not be expected to provide protection in the event rapid, targeted genomic mutations were to occur (as they already have in the last few months alone).
You might be wondering: if we can develop antibodies in vitro or transfer them from larger animals, what about using the serum of convalescent patients?
In fact, this was an approach proposed during the Ebola virus epidemic, but unfortunately, preliminary tests resulted in several dead animals.313 Essentially, this approach involves taking the serum of a patient that has recovered from COVID-19 and transplanting it into an infected individual. As has been stated multiple times in the medical and scientific communities that such a strategy is not only unproven but also carries certain risks.
For one, there is the obvious concern of transmitting other undetected infections that escaped pre-transfusion testing. Second, it may not be possible to find a large enough group of donors with high neutralizing antibody titers, and this may lead to accepting donors below the required threshold to meet demand. If this strategy were to enjoy a substantial share of clinical space, then desperate, hurried logistics would be almost a given.314
For this latter reason, if we cannot flatten the infection curve, the number of new cases will easily overrun available donors. And even if we had a sufficient number of donors, we could never guarantee the potency of each sample. That being said, we still do not know to what degree antibodies confer protections post-infection and whether re-infection could be possible, especially with antigenic shift and drift among viruses and mutations directly in the CoV-2 genome.
So while this approach may have limited ability to contain an ongoing outbreak, it could be a longer-term strategy for containing future outbreaks; especially if we implement proper follow-up testing post-infection.
Immunosuppressants
Synthetic pharmaceutical drugs with effects that mimic the hormones produced in the adrenal glands have long been used to suppress overactive immune systems. The most well-known are Dexamethasone, Prednisone, Florinef, and Hydrocortisone (cortisol). With respect to respiratory conditions, these drugs will most often be administered for severe asthma, COPD, hypersensitivity pneumonitis, eosinophilic pneumonia, interstitial lung disease, and sarcoidosis. The latter disease is actually a potential (but rare) complication of COVID-19 that involves excess accumulation of macrophages in granulomas.
Regardless of disease in question, past evidence with SARS and MERS would indicate that corticosteroids not only have zero effect on mortality rates, but also delay clearance of the virus. This would be particularly problematic if antibodies were being administered in parallel, for the reasons I stated above.315316
According to the World Health Organization, such drugs should not be routinely given systemically, and I would completely agree with this statement.317
Antimalarials
Perhaps the most well-known proposed therapy in association with COVID-19 is the antimalarial drug chloroquine. Currently, all the major hospitals in China, and to a lesser degree in other countries such as Korea and France, are either conducting clinical trials or reporting varying degrees of success. There has been a lot of mixed opinions and advice surrounding this drug, so more in-depth analysis of its beneficial effects as well as potential dangers are in order.
Therapeutic Benefits
Chloroquine Decreases ACE2 glycosylation
The most notable effect is one you likely haven’t heard of — chloroquine’s ability to decrease glycosylation of the ACE2 receptor, interfering with spike protein receptor binding. This fact was demonstrated as far back as 2005 with SARS-CoV, and though the receptor-binding domains of that virus and the new CoV-2 are slightly different, chloroquine would still be expected to have some inhibitory effect on receptor binding.318
Chloroquine prevents furin-mediated spike cleavage and inhibits inflammasomes
Chloroquine further shows the invaluable ability to inhibit or entirely prevent furin-mediated cleavage of the spike protein and release of viral RNA into the cell while at the same time inhibiting inflammasome activation.319 SARS-CoV is known to disturb intracellular potassium concentrations via viroporins, leading to inflammasome activation and secretion of the pro-inflammatory cytokines IL1B and IL18. As a matter of fact, any elevation in extracellular potassium will completely abolish inflammasome secretion of IL1B by inhibiting potassium efflux from cells.320
Another way in which virions activate inflammasomes is via lysosomal destabilization.321 Once the virion has been transported into the cell, encapsulated in a low-pH endosome, it will fuse with the host lysosome, forming an “endosome-lysosome complex”. The furin enzyme, which primarily resides in the trans-Golgi network, translocates to this newly formed endosomal-lysosomal compartment, where it proceeds to further cleave the spike protein (a process requiring low pH). This results in the fusion of the viral envelope and phospholipidic membrane of the endosome, providing an exit route for viral RNA into the cytoplasm of the cell.322323
Given that early endosome-lysosome compartments are rich in sodium ions, the combined lower endosome-lysosomal pH will cause further ionic disturbances by effluxing sodium, raising intracellular osmolarity. The cell attempts to accommodate by absorbing water through aquaporins, thereby leading to cellular swelling which, in turn, dilutes the potassium ion balance to below the threshold of 90 micromoles — the specific threshold which activates NLRP3 inflammasomes.324
It is by inhibiting lysosome/endosome acidification, thereby raising pH, reportedly by as much as 700-800 fold, that chloroquine is able to impair the release of the virus. Without sufficiently low acid, furin will not be able to cleave the spike protein for fusion with the endosomal membrane, and the virion will be unable to release its RNA into the cell for replication. Even for those virions that succeed in releasing their genomic material, chloroquine will act as a zinc ionophore, allowing extracellular zinc to enter the cell and inhibit RNA polymerase. Therefore, chloroquine, in addition to potentially blocking binding of coronavirus spike protein to ACE2 receptors extracellularly, also appears to have decidedly antiviral potential at the cell interior.325326327328329
Chloroquine mitigates granulomas and hypercalcemia
One of the complications of COVID-19 is a higher risk for sarcoidosis which, among other triggers, is associated with high blood calcium levels and elevated ACE and AngII.330 Sarcoidosis is characterized by the accumulation of macrophages into granulomas, typically surrounding bacterial or viral antigen. Though this disease is not frequently reported as a consequence of COVID-19, this may be in part due to the fact that macrophage accumulation and resulting fibrosis are common outcomes in ARDS, so it might be difficult to discern one from the other, especially as the severity of complications amplifies.331
One of the ways that sarcoidosis could be identified in the course of COVID-19 is by measuring blood and urine calcium levels — which are known to elevate due to the increased systemic expression of calcitriol (the active form of vitamin D) by macrophages.332 In patients with lung granulomas (which in COVID-19 would appear as alveolar macrophage clusters under high viral load), the most common biomarkers are hypercalcemia, hypercalciuria, and suppressed levels of parathyroid hormone.333 In fact, calcitriol directly regulates the production of several cytokines, decreasing proliferation of lymphocytes and monocytes.334
In this context, chloroquine has also been shown to reduce serum calcitriol levels in parallel with 24-hour urinary calcium excretion. This effect is attributed to its ability to inhibit the conversion of 25-hydroxyvitamin D (vitamin D3) to calcitriol, both in the kidneys and in macrophages directly.335
For those individuals supplementing with high doses of vitamin D3 and/or overexposing themselves to the sun, this will become an important consideration, especially if dietary calcium levels are high and/or vitamin K status is suboptimal.336
Chloroquine binds free heme
The next way in which chloroquine may provide therapeutic benefit is in its ability to bind heme. As discussed earlier, heme that has disassociated from globin and ferric iron can induce significant oxidative damage to both the vasculature and lung tissue. And while this would be an effect that would not be seen until later stages of ARDS when there could be higher degrees of hemolysis, the ability of chloroquine to bind heme, in concert with albumin and hemopexin, would be a notably valuable quality, especially in the absence of adequate hemopexin expression.337
Chloroquine inhibits macrophage activity
Chloroquine has also shown the ability to directly inhibit macrophage activity. By raising pH in endosomes, MyD88 and TRIM/TRAF-dependent immune pathways are disrupted, leading to lower activation of NF-kappaB. This, in turn, inhibits the degradation of IRF3, an important regulatory factor for antiviral interferon response. Chloroquine further dose-dependently up-regulates USP25 expression, a deubiquitinating enzyme responsible for clearing pathogens and misfolded protein Taken together, it is clear that Chloroquine will promote the uptake of viral pathogens into macrophages, thereby limiting their ability to provoke inflammatory processes.338
For many of the reasons listed above, medical experts have hastened to run clinical trials on chloroquine’s efficacy in the treatment of COVID-19. Unfortunately, there are several substantial dangers that must be considered if this therapy is to be implemented en masse.
Dangers of Chloroquine
Chloroquine induces CNS inflammation
Chloroquine has shown the ability to evoke inflammatory responses directly in the central nervous system by provoking astroglia to produce pro-inflammatory cytokines. This effect is primarily mediated by its ability to strongly elevate reactive oxygen species (ROS) by directly inducing the expression of NF-kappaB and chemokines CCL2 and IL8 in a dose and time-dependent manner.339
As a matter of fact, blocking NF-kappaB activation can completely abolish chemokine elevations during chloroquine treatment, which strongly suggests it is modulating their expression transcriptionally. Interestingly, chloroquine does not produce increases of ROS in experimental monocytic U937 cells, which may be induced to secrete large numbers of cytokines and chemokines. Therefore, it is presumed that the inflammation-enhancing effects seen may be specific to astrocytes and not a potential issue in monocytes / macrophages. Further, the inflammatory response was also absent in microglia.
For these reasons, it would appear that chloroquine produces ROS in astrocytes while at the same time suppressing immune responses in microglia, found in both the brain and infiltrating immune cells. That being said, given astrocyte implications in autism, schizophrenia, chronic pain, multiple sclerosis, and ALS, chloroquine use in people with these conditions should be handled with great care.
Chloroquine increases IL6
In another study looking at oxidative stress and lipid peroxidation in the liver as a result of fat accumulation, chloroquine was shown to increase liver enzymes (ALT / AST) and ROS, particularly IL6. Further, measures of TBARS showed significant elevations in lipid peroxidation. This effect is considered to be a result of chloroquine’s inhibition of autophagy due to disruption of lysosomes. In fact, impaired autophagy is associated with negative clinical outcomes in patients with non-alcoholic fatty liver disease (NAFLD). For this reason, chloroquine would be contraindicated in individuals with hepatic steatosis, NAFLD, or any other derangement of fatty acid metabolism.340341
Chloroquine lowers T and B cell proliferation
Chloroquine also appears to have some paradoxical influences over immune-modulation. On the one hand, it can inhibit inflammasome-mediated release of pro-inflammatory IL1B and IL18, as described above. Another study, however, demonstrated substantial suppression of IL2 and interferon-gamma, regardless of concentration. This could possibly indicate that its effect on inflammation may be more prominent than its direct antiviral capability. Nonetheless, ACE2 blocking would be of senior priority in evaluating the usefulness of chloroquine, in general, and we might forgive down-regulation of interferons by using adjunct therapies with opposite activity.342
Inhibition of inflammasomes is not the only way in which chloroquine can reduce pro-inflammatory cytokine levels. Due to its inhibition of acidifying maturation in endosomes, MyD88 and TRAM-TRIF-dependent pathways are affected as well, having systemic effects on other cytokines and chemokines. TRAM-TRIF, in particular, requires endosome activation, and is significantly impaired as endosomal pH rises. This results in lower IRF3 and, in turn, suppression of interferon response. This it one more way in which chloroquine may inhibit innate antiviral immunity.
Cardiovascular risks with chloroquine
In addition to the risks mentioned above, other dangers have been observed, including arrhythmias, prolongation of QT interval, tachypnea, tachycardia, hypoxemia, hypotension, and other EKG anomalies. Problems would only be exacerbated if Chloroquine were implemented in parallel with ACE inhibitors / ARB blockers, especially in the elderly with pre-existing cardiovascular disease. Such risks would extend to hydroxychloroquine as well.343
Conflicting results
In the field, there have also been conflicting reports. Chloroquine has proven effective against other coronaviruses, including SARS-CoV-1, but its effects on CoV-2 are still being evaluated.344345 Presently, there is insufficient evidence to fully embrace this drug as a therapeutic strategy, given some of the dangers I have listed above.
To date, in vivo studies have been limited to very small populations of people, and there have been a number of problems with their methodology, leading to conflicting results. For example, given that both chloroquine and hydroxychloroquine are metabolized in the liver and kidneys, any disruption to either organs could lead to complications.346347
Further, this treatment may be ineffective in individuals with porphyria (i.e. accumulation of cell-free heme structures), given its ability to bind heme. In such a context, it would be bound almost entirely by free hemes and not have the desired therapeutic effect. Further, patients with porphyria are also more likely to have pre-existing kidney stress.348
Unfortunately, there has been insufficient containment of public opinions regarding the safety of chloroquine, and it has led to the incidence of self-medication and overdose. Without knowing the medical history or genetic disposition of a patient, it is impossible to say whether chloroquine will be an appropriate treatment. For the public at large, the risk of liver and kidney toxicity increases with long-term use.349350351
The therapeutic dose of chloroquine phosphate (for malaria) has been set at 500mg, once a week, or 2.5g over 2 days. Nonetheless, deaths have been reported in children after ingesting just 1-2 tablets — doses as low as 300mg. Given that the lethal dose for adults has been estimated to be ~30-50mg / kg, that puts the death risk at ~2,000-3,400mg for a 150-pound individual. The lower end of that dose would comprise only four 500mg tablets.352
That being said, if a choice must be made, hydroxychloroquine (a chloroquine metabolite) has proven more soluble and less toxic than chloroquine, itself, producing fewer side effects. This does not ascribe full safety to its use, but nonetheless should factor into the therapeutic process when making decisions about which drug(s) to use.353354355
Antivirals
Antivirals are another option being considered, approaching the disease from another angle. The focus has primarily been on inhibiting the viral life cycle at different points in its process, with the main goal of blocking replication. Two broad targets have been identified: inhibition of viral polymerases and proteases. Inhibiting polymerases would block replication process directly, whereas protease inhibitors prevent proteolytic cleavage of viral protein precursors necessary for the production of new virion. Such targets have already been proven in models of HIV and Hepatitis C, however their applications for SARS-CoV-2 are still evolving.
At the moment, history seems to be repeating itself, as a great number of pilot clinical studies are in progress, assessing the potential of current antiviral medications for use against SARS-CoV-2. It should be noted that such a reaction in the scientific community has occurred in every single viral outbreak in history, and success has been extremely limited, outside isolated case reports. For example, none of the antivirals repurposed for use against Ebola were proven to improve patient outcomes. Nonetheless, here we are again amidst COVID-19, looking at many of the same drugs.356
Lopinavir-ritonavir drug combination
Lopinavir and Ritonavir, administered together, are a popular strategy and one of the most well-studied combinations. Being HIV protease inhibitors, it is thought they may have some clinical efficacy against SARS-CoV-2.357 Nonetheless, they have actually been demonstrated to increase chemokine CCL2 activity and IL6 secretion, both of which enhance inflammation. In fact, one study demonstrated that Pravastatin (a drug used to lower cholesterol) could inhibit this effect.358 Further, Ritonavir is decidedly hepatotoxic due in part to its inhibition of CYP3A4, a liver-expressed enzyme responsible for detoxification of numerous drugs and sex steroid hormones. For this reason, it carries a black box warning.359
One of the other unfortunate side effects of Ritonavir is its ability to block antigen presentation to CD8+ cytotoxic T-lymphocytes, thereby substantially lowering innate antiviral immune defense.360 Nonetheless, it has been proposed that Ritonavir could potentially activate natural killer cells via its down-regulation of MHC class I.361
At least one control study proved the Lopinavir-Ritonavir combination to substantially reduce adverse clinical outcomes during the SARS-CoV epidemic.362 Another placebo-controlled trial of the combination, with the addition of interferon beta-1b, was also used successfully among patients with MERS in Saudi Arabia.363 However, it should be fairly clear that this class of antivirals incur side effects that far outweigh their therapeutic benefits. For this reason, they are very weak candidates for the containment of COVID-19. One can only wonder why they are even being considered, given the higher safety profile of other therapies.
Remdesivir
This particular antiviral was originally developed by Gilead Sciences in 2017 for the treatment of the Ebola virus and has a broad anti-viral spectrum. It is an adenosine analogue which integrates itself into newborn viral RNA chains and results in their premature termination. Keep this property in mind, as I will be talking about another adenosine analogue, dipyridamole, in the section for curated compounds.
Remdesivir has already proven the ability to inhibit viral infection in human liver cancer cell lines, which are known to be sensitive to SARS-CoV-2.364 Other in vitro studies using epithelial cell cultures from primary human airway have demonstrated that Remdesivir can directly inhibit SARS-CoV and MERS-CoV replication.
Yet another study showed that Remdesivir in combination with interferon-beta was far superior to the Lopinavir / Ritonavir / Interferon-beta combination mentioned earlier.365 Therefore, it appears to be a more viable candidate for COVID-19 treatment.366 In fact, Remdesivir was used on the first COVID-19 patient in the United States, initiated on the evening of day 7. No adverse effects were noted and by day 8, his condition was already improving, including increased oxygen saturation values.367
Clinical trials for this antiviral have already involved 308 hospitalized adult patients with mild to moderate SARS-CoV-2 respiratory disease. The primary outcome was defined as a “time to recovery of 28 days”. This is the necessary elapsed time post-induction of Remdesivir treatment before we should see a reduction of fever, improved respiratory rate and oxygen saturation, and alleviation of cough for at least 72 hours.368
Only time will tell if this is going to be a viable ally containing the outbreak. Depending on results, it may need to be fast-tracked for rapid deployment in the event we see further mutations in this virus.
Antivirals considered safe for children
In general, the use of antivirals in children has been discouraged, given their potential side effects. Moreover, unless a child is severely immunocompromised, they are likely to have lower risk of severe outcomes, therefore not requiring such aggressive strategies.
IFN-alpha
If antivirals are to be used at all in this population, perhaps the safest alternative would be interferon-alpha (IFN-alpha), which has shown the ability to inhibit the synthesis of viral RNA and replication. Though it is produced by the body in response to viral infection, it is likely to be suppressed at later stages of disease progression. In fact, it is one of the cytokines that is insufficiently expressed in the immunocompromised.
In China, IFN-alpha is available in injections, sprays, and gels and is commonly included in nebulizers.369 In fact, proposals have been made that IFN-alpha spray be dispensed to families or groups of people with a history of close contact with COVID-19 infected patients.
Caution should nonetheless be exercised, however, given that IFN-alpha may increase liver enzymes, induce kidney damage, and promote excessive bleeding. For this reason, IFN-alpha is contraindicated for patients with abnormal liver function, including those with lower creatinine clearance.
Ribavirin
As another safer option for children, Ribavirin also has inhibitory effects on both RNA and DNA viruses. In the USA and Europe, it is available in both oral and inhalant forms for children 3 years and older. For children with active COVID-19 infections, Ribavirin injections of 10 mg/kg (maximum 500mg), 2-3 times daily has been recommended.370
As with all antivirals, in general, this drug is contraindicated in individuals with abnormal liver function. Another potential side effect of Ribavirin is its ability to enter and accumulate in red blood cells, potentially leading to hemolytic anemia. For the reasons I have stated above, increased hemolysis should be avoided at all costs during the course of this disease, given the risk for cell-free hemoglobin-mediated damage to airway epithelial cells and the vasculature in general.371 Therefore, if Ribavirin is to be considered in any context, it is mandatory for hemoglobin and hematocrit levels to be monitored closely.
Arbidol
Last but not least we have Arbidol, which has proven to effectively inhibit SARS-CoV-2 at a concentration of 10-30 micromoles — in vitro.372 This antiviral has been used for some time in both Russia and China for the treatment of influenza, but is less widely known in the USA and Europe. There is currently no Arbidol dosage recommendation for children in COVID-19, but the same precautions apply for liver toxicity.
Taken together, some general advice may be offered regarding the use of antivirals, particularly in children.
- If symptoms are mild, a low dose of IFN-alpha via nebulization may be all that is required to resolve the condition.
- Ribavirin may be used as an adjunct to other therapies, but would likely not be effective alone.
- No more than two antivirals should ever be used at the same time, due to the combined, amplified risks of liver toxicity.
Other antivirals
A few other lesser-known options are currently being considered, and may work well as adjuncts in multi-pronged therapies that also modulate inflammation. They include the anti-HCV nucleotide inhibitor Sofosbuvir and protease inhibitor Rupintrivir.373374
Camostat
The third contender is the serine protease inhibitor, Camostat, which may be the most promising option of all. Originally approved in Japan for the treatment of chronic pancreatitis and postoperative reflux esophagitis, this drug has also shown the capability to inhibit TMPRSS2-mediated cellular entry by SARS-CoV. Further, at least one in vitro study has shown that Camostat can significantly reduce the infection of Calu-3 lung cells by SARS-CoV-2.375376377378
It’s profile is more favorable than other compounds we have discussed thus far, as it has also demonstrated the capability to inhibit fibrosis in liver or kidney disease. This characteristic may very well extend to pulmonary fibrosis as well, given that the primary mechanism by which Camostat prevents fibrosis is by inhibiting expression of chemokine CCL2, which is responsible for neutrophil infiltration into the lungs of ARDS patients.379
It would appear that derivatives of Camostat have also been developed to enhance its immuno-modulating properties. For example, ONO-3403 significantly inhibits TNF-alpha and immune-induced nitric oxide production. ONO-3403, further, has even stronger protease-inhibitory activity, making it another contender for a viable COVID-19 therapeutic.380 Moreover, Camostat has also been investigated as an anti-hypertensive.
 Antibiotics
Antibiotics
Antibiotics have been considered primarily for their anti-inflammatory effects, given they have no proven antiviral activity. Therefore, they are often given in conjunction with one of the other compounds we have discussed, particularly antimalarials.
Dactinomycin
Dactinomycin, also known as actinomycin D, in a member of the cytotoxic antibiotic family of medications and is frequently used in chemotherapy. It his highly suppressive of hematopoietic stem cell differentiation in bone marrow and can even induce cancer, allergic reactions, and necrosis of tissue at the site of injection. Nonetheless, it may surprise you that it has been approved for medical use in the United States since 1964 and enjoys a place on the World Health Organization’s “List of Essential Medicines”.381382
The list of side effects, alone, for this drug would turn away even the least conservative of medical professionals. Nonetheless, it has been formally proposed for use in the fight against COVID-19 — unfortunately, based on nothing more than a computer simulation.383 This is a perfect example of how network-analysis of the “interactomes” of compounds, genetics, and immunological pathways can lead to disastrously wrong conclusions. Does this mean that such “Big Data”-sourced analytics are not useful for identifying potential drug candidates? The answer is an obvious “no”, however I should mention that without proper human oversight and involvement of immunologists, virologists, and other qualified specialists, such analytics are prone to erroneous conclusions — as we have already seen with the “hemoglobin attack myth”.
Nonetheless, Dactinomycin does seem to have an interesting profile which includes the promotion of angiotensinogen (AGT) inhibition by Spironolactone, Valsartan, and Amiodarone,384385 broad immunosuppression resulting in higher protection against vascular injury and permeability,386387388389390 and, paradoxically, inhibition of virally-induced IL10 (which itself is immunosuppressive).391
Does this mean it can be safely used in the context of COVID-19 treatment? A better question would be — would you use chemotherapy to kill a virus? That is essentially what this “computer-model generated” suggestion would have you do.
Azithromycin
This antibiotic may be a better option, and medical authorities are giving it closer attention. It’s predominant function is antimicrobial activity against secondary bacterial infections resulting from primary SARS-CoV-2 viral infiltration.392 It has also been shown to have decidedly anti-inflammatory effects as well as inhibiting IL1B in alveolar macrophages,393394 decreased expression of IL6 and TNF, and lowered CCL2-mediated neutrophil infiltration.395
Despite these positive attributes, it does come with a handful of side effects including QT prolongation and arrhythmia, liver toxicity, and exacerbation of myasthenia gravis. There is also the potential for significant disruption to the gut microbiome, which could have further negative immune-modulating outcomes. Therefore, for the majority of people, this strategy should only be considered if symptoms are resistant to other approaches with a higher safety profile.
Teicoplanin
This glycopeptide antibiotic may be the unsung hero, in that it has already shown activity against previous viruses such as Ebola, influenza virus, flavivirus, hepatitis C virus, HIV virus, and even MERS-CoV and SARS-CoV.396 It is routinely used to treat bacterial infections such as gram-positive Staphylococcus, and has been added to the anti-COVID-19 arsenal due to its potential ability to block SARS-CoV-2 cell entry.397
According to Zhou et. al.398, teicoplanin works very early on in the viral cycle by inhibiting the low-pH cleavage of the viral spike protein, thereby preventing release of genomic viral RNA and subsequent viral replication — an effect also demonstrated by chloroquine.
Although it was proposed by Zhou that cathepsin L is the primary endopeptidase responsible for spike protein cleavage, furins have also been implicated, as I have mentioned previously. Nonetheless, teicoplanin does not associate with either cathepsin L or furin and, like chloroquine, directly inhibits acidification of the endosome-lysosomal compartment Moreover, 1.66 micromoles of teicoplanin is all that is needed to inhibit 50% of viruses in vitro — a level much lower than that reached in human blood on a typical 400mg dose.
Though further randomized clinical trials will be required to confirm these preliminary results, this antibiotic does seem to have a far greater safety profile than the others being considered now.
RAS Modulation
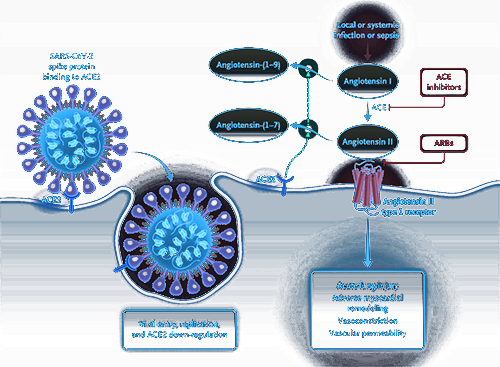 I have already discussed at length the renin-angiotensin system and its implications for cardiovascular complications in COVID-19. It should come as no surprise that this system is another major target for COVID-19 therapies. From my perspective, however, the full implications of RAS inhibition — and ACE2 in particular — have not been thoroughly considered, and the preponderance of attention has been placed on ACE2 receptor blocking. For a wide variety of reasons, this could lead to unexpected consequences.399
I have already discussed at length the renin-angiotensin system and its implications for cardiovascular complications in COVID-19. It should come as no surprise that this system is another major target for COVID-19 therapies. From my perspective, however, the full implications of RAS inhibition — and ACE2 in particular — have not been thoroughly considered, and the preponderance of attention has been placed on ACE2 receptor blocking. For a wide variety of reasons, this could lead to unexpected consequences.399
To briefly re-summarize, the RAS system regulates electrolyte balance and blood pressure by first producing angiotensinogen (AGT) in the liver. Angiotensinogen is secreted into circulation where it is cleaved to angiotensin I by kidney-derived renin. The angiotensin-converting enzyme (ACE) then further cleaves it down to the octapeptide Angiotensin II, which exerts a wide array of pathological effects via its receptor, AT1R.400401
As I have detailed above, AngII activation of AT1R has multiple effects, not the least of which is weak bronchoconstriction, activation of NF-kappaB and increased pro-inflammatory cytokines, increased NADH / NADPH oxidase to produce ROS, and the direct activation of Toll-like receptor 4, inducing apoptosis and lung fibroblast proliferation. Activation of AT2R, however, is a completely different story and actually has just the opposite effect by promoting vasodilation and inhibiting growth signals that lead to fibrosis.402403
Apart from the conversion of AngI to AngII, the ACE enzyme itself has other actions that include the degradation of bradykinin and substance P, two pro-inflammatory peptides that can trigger the release of prostaglandins and nitric oxide, leading to cough. ACE2 is co-located in cell membranes with AT1R and AT2R and actively lowers blood pressure by degrading AngII into vasodilatory angiotensin (1–7). As such, there is delicate cross-talk between them that leads to varying degrees of pro-inflammatory dysregulation.
Currently, ACE inhibitors and AT1R blockers are being proposed for new COVID-19 patients, given that elevated ACE and AngII are prognostic factors for severe pneumonia. Further, SARS-CoV-2 induces the shedding of ACE2, thereby lowering its ability to modulate the RAS system and lower blood pressure. For this reason, blocking ACE2 to prevent viral entry will have the simultaneous effect of increasing risk for hypertension, thrombosis, and eventually — vascular permeability and hemolysis.
ACE inhibitors and ARBs
ACE inhibitors were first proposed following the SARS-CoV epidemic of 2002-2003, given that animal experiments had shown RAS inhibitors could relieve symptoms of acute severe pneumonia and respiratory failure.404405 One of the prominent side effects of this therapeutic approach, however, is a persistent, dry cough.406 Nonetheless, the medical establishment stuck with the original recommendations and continue to prescribe ACEIs , as they had shown the ability to decrease mortality rates.407 For this reason, such drugs are currently being proposed as treatment for COVID-19 patients in the ICU.
Angiotensin receptor blockers (ARBs) have also been considered for COVID-19, albeit working in a different manner. Instead of inhibiting conversion of AngI to AngII, they block AngII binding to ATR1 receptors. AT1R antagonists such as Losartan and Olmesartan, commonly used to treat hypertension, have demonstrated the ability to potently increase ACE2 expression in cardiac tissue (by up to three-fold) after chronic treatment for 28 days, reducing mortality rates from cardiac infarction.408
Modulating the RAS system is not without clinically opposing effects. The AT1R antagonist, Losaratan, for example, has been shown to up-regulate renal ACE2 expression. Higher urinary ACE2 levels are one of the indicators of hypertension.409 The general consensus is that chronic AT1R blockage leads to an up-regulation in ACE2 expression.
As mentioned earlier, this is believed to be a problem in COVID-19, given that SARS-CoV-2 binds to ACE2 on host cell membranes in order to gain entry. At first glance, it might seem counterintuitive to elevate ACE2 expression in COVID-19 patients using an AT1R antagonist. However, if we turn to the SARS-CoV (2002-2003) studies, spike protein binding to ACE2 eventually leads to ACE2 down-regulation. The result is an accumulation of of AngII, leaving ACE2 scrambling to convert it to the vasodilating Ang(1-7). In reality, with increasing viral loads, ACE2 levels will quickly plummet, and the patient will be left with excess AngII and a potential hypertensive crisis.
It is specifically this cascade that leads to acute lung injury, given AngII’s ability to increase vascular permeability via AT1R.410411 For this reason, it would appear that the higher ACE2 levels seen in COVID-19 patients medicating with AT1R blockers are actually protecting them from acute lung injury rather than putting them at higher risk for SARS-CoV-2 infection!
ARBs afford this protection via two complementary mechanisms:
- They prevent excessive activation of AT1R induced by viral infection and
- Due to the up-regulation of ACE2, they reduce AngII accumulation and increase the vasodilator Ang(1-7).
Losartan: Friend or Foe?
Losartan is the most well-studied ARB and therefore the more popular candidate in COVID-19 therapeutic strategies. By preventing AngII binding to AT1R, it side-steps the pathogenic effects of excess AngII activity.
Losartan inhibits NADPH oxidase
One such AngII-associated activity is the generation of reactive oxygen species (ROS). In the course of viral infection, AngII directly activates NADPH oxidase via AT1R, leading to fibroblast growth and collagen synthesis.412413414
It is this activation of NADPH oxidase, in particular, that appears to be a key driving factor in pulmonary fibrosis. NADPH oxidase catalyzes the generation of superoxide, thereby increasing vascular permeability. In the brain, this also results in injury to the blood-brain barrier.415 Therefore, Losartan acts as a “causative” antioxidant by preventing AngII induction of superoxide release.416
Losartan decreases chemokine expression
Its effects do not stop with protection from oxidative stress. Elevated chemokine expression is associated with negative clinical outcomes due to increased infiltration of immune cells to sites of infection. Losartan in lower doses of 5mg / kg has also proven to directly decrease CCL2 expression, thereby inhibiting monocyte/macrophage infiltration into lung tissue.417 In fact, it has been shown that endothelial cells interact with granulocytes primarily through AT1 receptor — a factor that has very compelling implications for pneumocytes.418
Losartan inhibits inflammation and lung remodeling
AngII has also been shown to stimulate AT1 receptors directly on macrophages, further enhancing their inflammatory potential.419420 As respiratory distress proceeds, increased hypoxia induces the expression of adhesion molecules periostin (PN) and osteopontin (OPN) in pulmonary arterial smooth muscle cells.421 This leads to fibrosis, characterized by pulmonary vascular hypertrophy and remodeling.
AngII in the lung has shown to be a potent stimulator of both PN and OPN expression. Bronchial epithelial cells and alveolar macrophages in even normal lungs express OPN, the overexpression of which is associated with a large array of lung diseases.422423 In fact, OPN interacts with multiple, ubiquitously expressed cell surface receptors and plays extraordinarily varied roles ranging from wound healing to inflammation, ischemia, and immune responses.
In the last decade, OPN has also been associated with allergy and asthma. In these conditions, OPN plays a key pro-inflammatory role during the sensitization to allergens. Of note, OPN deficiency has even been reported to protect against airway remodeling.424 By blocking AT1R receptors, Losartan may further prevent AngII from stimulating PN and OPN, thereby mitigating pulmonary fibrosis.
Losartan promotes vasodilation
One of the other effects seen with AngII is its ability to increase vascular sensitivity to norepinephrine, thereby inducing vasoconstriction. This effect would be exacerbated by elevations in any of the co-factors for norepinephrine production (e.g. copper and ascorbic acid).425 To add insult to injury, AngII further diminishes re-uptake of norepinephrine in renal sympathetic nerves, increasing renin output and, thereby higher AngII levels, creating a vicious circle.426
By blocking AT1 receptors, ARBs reduce vascular reactivity to norepinephrine. In doing so, it can dose-dependently decrease renal vasoconstriction response to sympathetic nerve stimulation.427428429 This is especially important in the vasculature of the kidney, which is exquisitely sensitive to fluctuations in blood pressure.430
Females are more vulnerable
Unfortunately, it would appear that females (especially pre- and peri-menopausal) and men with higher estrogen levels are at elevated risk for AngII-mediated hypertension due the ability of estrogen to alter pituitary response to AngII.431432 Estrogens have long been known to enhance AngII biosynthesis, in part by increasing angiotensinogen mRNA expression in the pituitary.433 AngII release directly from the hypothalamus has also been demonstrated, in response to progesterone and estrogen.434
An ally against ARDS
All these factors taken into consideration, it’s clear how important RAS regulation is in the pathogenesis of ARDS.435436437 RAS components are found not only throughout the entire vascular system but also directly in lung fibroblasts, alveolar macrophages, and airway epithelial cells. Further, each of these cell types are capable of directly synthesizing the RAS peptides.438
One of the other ways that Lorsartan may protect against vascular injury, permeability, and ultimately — hemolysis — is by directly stimulating the production of nitric oxide.439 The less endothelial nitric oxide there is, the greater potential for vasoconstriction and, as I hope I’ve made abundantly clear, hemolysis and cell-free hemoglobin are a non-issue if hypertension and increased vascular permeability may be avoided.440
Activated granulocytes have shown the ability to produce superoxide, thereby inactivating endothelial nitric oxide. Such eNOS inhibition actually increases their adherence and migration, thereby magnifying microvascular permeability.441442 In fact, any inhibition in the production line for NO will increase basal permeability of peripheral microvessels, especially those surrounding the alveoli.443444 Not only can Losartan indirectly improve nitric oxide levels, via its effects on the RAS system, but it has also been shown to prevent stroke and decrease the incidence of edema and ischemia.445446447
Other therapeutic benefits
There are yet other ways that Losartan could benefit patients with COVID-19 ARDS. Losartan also increases vascular and cardiac HGF levels, further activating eNOS expression and conferring further cardiovascular protection.448449450451
ACE2 monoclonal antibody
For reasons that should be obvious at this point, monoclonal antibodies to ACE2 for the purposes of driving down virion entry into cells could produce very undesirable results, not the least of which is loss of regulatory control over AngII levels. Further, without modifying Fc receptors in the antibody product, there is a much higher potential for unexpected inflammatory responses in any and all tissues expressing ACE2.452453 Such necessary modifications would, of course, increase the expense for producing the antibodies, further prolonging the turnaround time for much-needed therapies. Nonetheless, they would be mandatory, from my perspective, as they afford the clinician a longer half-life, allowing other strategies to be implemented in parallel for more rapid recovery.
I personally do not believe, given what we know about ACE2 and AngII interactions, that monoclonal antibodies are a sustainable strategy for the containment of this virus or any of its future progeny / family. ACE2 shedding happens as a natural result of increased viral load, and the issue is less about preventing cell entry and more about depriving the virions of their ability to hijack ribosomal machinery and induce inflammasome-mediated inflammation.
ACE2 immunoadhesin
The immunoadhesin strategy would bind to the virion itself rather than to an ACE2 receptor on the host cell. A soluble version of the ACE2 receptor capable of binding to the spike protein has been proposed. Such a strategy has proven capable of blocking SARS-CoV from infecting cells — but unfortunately, this effect has only been demonstrated in vitro.454 The theory wasn’t tested in animal models, though a previous study indicated that 50% inhibition could be attained with a complex consisting of an ACE2 extracellular domain with a human IgG1 domain.455
This approach seems to be far more tenable, especially considering that it does not block ACE2’s much-needed AngII-degrading function and prevents ACE2 shedding by binding to the spike protein and preventing attachment. This strategy could be made even more effective if it were to be administered along with recombinant ACE2 protein, to make up for concurrent shedding induced by existing virions. Such an approach could possibly be the best of both worlds.456
As a matter of fact, clinical trials of recombinant ACE2 have shown that it is well tolerated.457 In this scenario, even if SARS-CoV-2 were to attempt to circumvent ACE2 neutralization by decreasing receptor affinity, in doing so, it would essentially become less pathogenic. In fact, this is precisely why SARS-CoV was less pathogenic — due to a lower ACE2 binding affinity.458
Taken together, it could be that the most effective therapeutic strategy could involve ACE2 immunoadhesins combined with recombinant ACE2 and low-dose angiotensin-receptor blockers. The latter adjunct would be clearly contraindicated in those with low blood pressure or patients in ICU with hypotensive sepsis. Nonetheless, the majority of acute COVID-19 cases have just the opposite profile with vasoconstriction, acute pulmonary inflammation, and hypertension. In this context, RAS-system modulation perhaps makes the most sense.
Anti-fibrotics
Betulinic acid
Betulinic acid is the active ingredient isolated from the bark of the birch tree Betula spp. (Betulaceae) It may be a possible adjunct in COVID-19 therapies via its potent attenuation of hydroxyproline and smooth-muscle actin. Though this action has been demonstrated in liver, it would be expected to operate similarly in lung. Indeed, it has already shown the potential to reduce neutrophil recruitment and, thereby, lung inflammation in models of bacterial infection.459
It appears to reduce inflammation, in part, by inhibiting NF-kappaB activation. In parallel with its inhibition of metalloproteinase-1 and MMP-13, it should be presumed that it might be a powerful ally in the phases of this disease where there is pneumocyte hyperplasia and lung remodeling.460
Novel Therapies: On the Cutting Edge
 The Problem with Predictive Models
The Problem with Predictive Models
At Transcend Genomics, we use gene, pathway, and compound networks to derive customized therapeutics for our clients. As such, we know the pitfalls of relying exclusively on data generated with algorithms, even those enriched with machine learning / AI. It seems that such network-type analysis has become quite popular since the onset of COVID-19, given the limitations of existing medicines in addressing its unique challenges. Drug targets within cellular networks have been coupled with therapeutic profiles; however, there are inherent problems with this approach, given the inherent lack of real-world testing. Similar to the “hemoglobin attack” myth that has unfortunately swept the world over the last several months — convincing many people of SARS-CoV-2’s capability of directly dissociating iron from hemoglobin — predictions for novel therapies could likewise mislead unsuspecting researchers less familiar with the nuances of molecular biology and virology.
A good example of how predictive analytics can go wrong is a recent network-driven analysis from China published in Nature this past March. They presented very impressive “interactome networks”, though the drug that revealed the highest efficacy for CoV treatment was Mesalazine — a compound capable of inducing pneumonia and lung damage.461462463
Interactomes aside, some interesting proposals have arisen over the past several months, partly driven by the medical record, partly informed by predictive analytics. Though many of them have not yet been tested, they should nonetheless be considered in order to expand our potential arsenal for COVID-19 therapeutic strategies.
Recombinant Haptoglobin / Hemopexin
Iron overload and/or excessive cell-free hemoglobin resulting from hemolysis has not yet been demonstrated in COVID-19 ARDS, despite what “hemoglobin attack” proponents are saying now. Nonetheless, haptoglobin and hemopexin-based therapies could be useful in containing complications in ARDS, especially in those cases where there is excessive vascular permeability. While hemopexin can scavenge cell-free heme, haptoglobin, in concert with CD163-expressing macrophages, may clear cell-free hemoglobin.464465
Unfortunately, administration of recombinant haptoglobin and / or hemopexin would be minimally effective in situations where there has already been extravasation of reactive cell-free heme into the extravascular space. This is due, in part, to the fact that hemopexin cannot efficiently prevent accumulation of heme extravascularly, as it rapidly intercalates into lipid membranes, inaccessible to circulation.
Therefore, it’s important that such therapies be implemented prior to widespread vascular damage, thereby allowing haptoglobin to prevent oxidation of oxyhemoglobin and bind / sequester it from circulation. Such rationales have supported the therapeutic use of both haptoglobin and hemopexin.466467 Nonetheless, depending on the degree of hemolysis, such scavengers could be depleted quite rapidly, leading to quick reactions of oxyHb with nitric oxide, further driving vasoconstriction and increased vascular permeability.
Earlier studies that argued the need for reducing agents to keep cell-free hemoglobin in its reduced ferrous-state (i.e. oxyhemoglobin) failed to appreciate the volatile dynamics of nitric oxide, haptoglobin, and hemopexin, all of which could become dysregulated, depending on multiple factors.468
Pre-clinical proof-of-concept animal models have shown that plasma derived human haptoglobin and hemopexin can attenuate vascular damage from cell-free hemoglobin and heme.469
Given that cell-free hemoglobin is filtered through the kidneys once haptoglobin has been depleted, we are clearly on the razor’s edge with this therapy. Not enough, and there will be comprehensive oxyhemoglobin-NO reactions leading to hypertension. Further, haptoglobin demand increases in parallel with antioxidant repletion (note, not depletion), due to increased reduction of methemoglobin to oxyhemoglobin. Balance is everything here.470
Anti-cancer / Anti-inflammatory
Cepharanthine
Cepharanthine is an isolate extracted from the Stephania cepharantha Hayata plant, a native to eastern and southern Asia and Australia. It has been approved in Japan for the treatment of alopecia areata and as an adjunct to radiation therapy to prevent anemia and thrombocytopenia. It is not very well studied, but nonetheless seems to have some desirable properties that may be useful in models of autoimmunity and allergy.
The most relevant effects for this compound are as follows:
-
Inhibits lipid peroxidation / stabilizes membranes471472473
-
Anti-malarial474475
-
Anti-inflammatory476
-
Inhibits platelet aggregation477478479
-
Scavenges free radicals480481
Its primary mechanism of action is allegedly as an inhibitor of the efflux transporter ABCC10, with secondary binding to heat-shock protein Hsp90. A follow-up in the Chinese Medical Journal shows some potential CoV antiviral activity, but it cannot be determined from existing data whether or not this drug could be used as a potential therapeutic strategy. Further investigations, optimally with human clinical trials, would be necessary before implementing it in a formal clinical regimen.482
Anti-inflammatory cytokines
Another option for managing widespread inflammation caused by SARS-CoV-2 is to implement recombinant human anti-inflammatory cytokines such as IL37 or IL38.483 CoV-2 stimulates these cytokines, often in inadequate amounts, in response to lung inflammation, fever and fibrosis. Therefore, using them as a potential therapeutic strategy may be relevant.
IL37, a member of the IL1 family, suppresses innate and humoral immune responses via its interactions with ILR5 and IL18Ra receptors.484 It has been shown to suppress expression of pro-inflammatory IL1B, IL6, TNF-alpha, and chemokine CCL2.485 It’s inhibition of IL1B, in particular, has lead many to believe that it could potentially dampen inflammasome mediated immune response.
IL38 also suppresses inflammation, with IL1, IL17, and IL22 as its primary targets. It is produced primarily by B cells and macrophages in response to virally-induced inflammation. The risk with using such a therapeutic approach would be the possibility of over-suppression of pro-inflammatory immune factors, losing proper balance of innate and humoral immune responses. Knowing the patient’s immune status and current stage of disease would be mandatory.
SERMs and estrogenic steroids
On the other side of the spectrum we have selective estrogen receptor modulators (SERMS), which leverage estrogen’s ability to influence immune function. Overexpression of the estrogen receptor has been shown to inhibit viral replication, and SERMs have been considered as mechanisms to duplicate this effect.486
Toremifene
Toremifene, the first generation of nonsteroidal SERM, has been considered for its ability to block viral infections, including MERS, SARS, and Ebola viruses inside cells.487488
It works by preventing fusion between viral and endosomal membranes, interacting with and destabilizing the virus membrane glycoprotein. This eventually leads to an inhibition in viral replication.489
Nonetheless, use of SERMs is limited by the potential estrogen receptor activation might have for induction of angiotensinogen expression in the liver. For this reason, their risk for AngII enhancement may be real.
mTOR inhibitors
The mammalian target of rapamycin (mTOR) is a kinase that coordinates, via interactions with other proteins, cell growth, proliferation, motility and survival. It also plays a major role in protein synthesis, autophagy, and transcription.
mTOR is comprised of two distinct complexes (mTORC1 and mTORC2). mTORC1, in particular, senses deficits in nutrients / energy and responds by up-regulating protein synthesis. mTORC1 is regulated primarily by insulin, growth factors, phosphatidic acid, certain amino acids and their derivatives, mechanical stimuli, and oxidative stress.
mTORC1 has further been confirmed as a key factor regulating various viral replications.490491
Sirolimus
Sirolimus (also known as rapamycin), an mTOR inhibitor, is used for its immunosuppressant functions, especially in the context of kidney transplant rejection. It primarily works by reducing T and B cell sensitivity to IL2 stimulation, thereby inhibiting their activation. It has also shown the ability to effectively block viral protein expression and subsequent release.492
Sirolimus has further shown potential to improve clinical outcomes when used in the context of H1N1 pneumonia and acute respiratory failure. As with any immunosuppressant, there is a fine line between too much and too little — and I would imagine that line could easily be crossed, depending on what stage of COVID-19 a patient is in. For this reason, broad immunosuppression, especially of lymphocytes in a context where late stage infections show lymphopenia — is not likely to be the best idea. In the case of B cells, inhibition could easily blunt antibody response, which would only be a desirable outcome in individuals at risk for antibody-mediated inflammation, as I have described earlier.493
Oligonucleotides
Oligonucleotides are short DNA / RNA sequences (i.e. oligomers) that have been applied in a wide variety of contexts from genetic testing to forensics. There is a possibility they could be used to directly target CoV-2 surface proteins or the RNA genome for rapid degradation. For example, small interfering RNA (siRNA) or antisense oligonucleotides (ASO) might be used to target viral RNA genomes.494
In order for the strategy to work, the oligonucleotides would need to be delivered directly to the lungs. There are some recent technological advances such as lipid nanoparticles that could serve this purpose, but they have not been tested in this context. Antigenic reactions are highly possible.495
Another reason such an approach could fail is because of the large surface area for alveolar epithelial cells and variability in viral load. It might be quite difficult to ensure sufficient levels of oligonucleotide were present in the lung to cover all surfaces. This is probably one of the reasons that siRNA’s did not prove effective with Ebola.496
Further, siRNA / ASO therapeutics are currently only considered for rare diseases, so there’s a big unknown as to whether or not we could sustain a mass production line for the public at large.
RBD to Fc fragment
Nanobody / VHH domains
Another potentially novel therapy would involve single-domain antibodies (sdAB or “nanobodies”). These are antibody fragments that, similar to a complete antibody complex, are able to selectively bind to antigens. These are attractive due to their size, which is over 10x smaller than whole antibodies.
The first nanobodies, called VHH fragments were engineered from heavy-chain antibodies found in camelids (from the camel family). Such VHH fragments have been shown to be equally as effective as whole structures and even more resilient. Further, they are easily cultivated in vitro, making them a more alternative option than the other antibody approaches we have already discussed.497498
VHH fragments could rapidly diffuse systemically but their half-life would be decidedly short due to the absence of an Fc domain.
 COVID-19 Natural Approaches: Can They Work?
COVID-19 Natural Approaches: Can They Work?
Given the severity of the COVID-19 epidemic, there has been a surge of interest in vitamins, supplements and other natural therapies that could protect against infection and boost immune resilience. Unfortunately, this epidemic has also attracted a large number of opportunists looking to promote one or another “silver bullet”, making often wild and irresponsible claims ranging from antiviral activity to even reversal of acute respiratory distress.
Many, if not the majority, of these suggestions are being made by chiropractors, doctors of functional medicine, and nurse practitioners, though there has also been an upsurge of activity among independent researchers with often less than complete “foundational” education in molecular biology and immunology. In some cases, conclusions have been drawn that could potentially hurt individuals sincerely looking for answers. Therefore, it’s of critical importance to discern fact from fiction in a growing ocean of information. Some of the suggestions that have surfaced do have merit, but they must be taken in context and understood with full consideration of the stages of disease and pathological implications described in detail above. In my opinion, such subtleties have been side-stepped in many cases, and this can lead to clear danger for the public at large.
 Emodin
Emodin
The first natural compound I would like to discuss is neither well known nor actively promoted in mainstream health circles, though it has been a component in Chinese medicine for years. It could be a major player in the fight against rapidly mutating viral threats such as SARS-CoV-2.
Emodin is a chemical isolated from rhubarb, buckthorn, and Japanese knotweed. It is commonly found in many traditional Chinese medicines and is known, first and foremost, by its anti-inflammatory effects. Of interest to the clinician is its antiviral activity against coronaviruses, in particular SARS-CoV-2.499
Of greatest interest is Emodin’s demonstrated ability to dose-dependently prevent binding of the spike protein to ACE2 and directly inhibit ORF3a protein activation of the inflammasome.500501
Inhibits vascular remodeling
It’s effects do not end at ACE2, as it also inhibits vascular remodeling by arresting vascular smooth muscle cell proliferation.502
A powerful antioxidant
Emodin has also proven to be a powerful antioxidant, acting as a potent free radical scavenger, powerfully attenuating concentrations of IL1B, IL6, and TNF-alpha in bronchoalveolar lavage fluid.503
Further, emodin enhances heme oxygenase expression, further increasing potential for heme catabolism — a useful effect in contexts where higher levels of cell-free hemoglobin are present (e.g. in late-stage ARDS).
This enhancement is partly attributed to activation of transcription factor NRF2, which is critical for protecting the lungs against oxidative stress. NRF2 directly increases expression of antioxidants such as glutathione peroxidase and glutathione reductase.504505506 Emodin apparently directly promotes NRF2 activity, making it a potentially valuable ally in the protection against lung injury.
Emodin has also shown strong protection against lipid peroxidation, caused by either neutrophILderived oxygen radicals or from cell-free heme. Malondialdehyde (MDA) is one of the most common indicators used to gauge lipid peroxidation, and Emodin can significantly lower MDA and raise superoxide dismutase (SOD), thereby inhibiting oxygen-free radical damage.507508509
Reduces NF-kB nuclear translocation
NF-kappaB is the primary transcriptional regular for pro-inflammatory cytokines and has been strongly correlated with the axis of inflammation, fibrosis, and cancer.510
Emodin has also shown the ability to directly reduce nuclear transcription of NF-kappaB. This could have far-reaching implications for virally-overactivated inflammasomes.511
 Melatonin: Wolf in Sheep’s Clothing
Melatonin: Wolf in Sheep’s Clothing
This pineal hormone needs no introduction, and it has been positioned front and center in the fight against COVID-19, given its history of use in the context of viral infection. Unfortunately, it’s effects on the immune system are not as straightforward as many would have you believe.512513
Melatonin was one of the natural remedies proposed during both the SARS-CoV and Ebola virus epidemics and it was touted to have a virtually pristine safety profile, working both as an immunomodulator and an antioxidant. In the case of SARS, it was even shown to target several keys genes such as ACE2.514515516
It is melatonin’s antioxidant effect, in particular, that make it such a strong candidate for antiviral treatment, regardless of the fact that melatonin has not been proven capable of curbing viral replication or lowering viral load.517
Nonetheless, it’s varied effects on inflammation have made it an attractive option for prolonging survival time and thereby widening the recovery window for individuals with weaker immune systems. But therein lies the rub: the immune status of the patient can determine melatonin’s effects, and it has a very clear potential to take a detour into very undesirable territory.
Melatonin is a sleep-regulating hormone produced by the pineal gland in the absence of light, and blue light, primarily in the 460-480nm wavelength (as found in bright sunlight) suppresses its secretion. Its potent antioxidant and free radical scavenging ability was first observed in 1993 at the University of Texas. It has potential actions on melatonin receptors at locations far beyond the brain, throughout the entire body, promoting antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase.518519
Exceptionally high levels of melatonin have been found in mitochondrial fluid at much higher concentrations than that found in blood plasma. This has led to the conclusion that it is one of the primary mitochondrial antioxidants.520521522 That being said, robust clinical studies surrounding melatonin’s efficacy in disease treatment are few and far between and its potentially paradoxical effects on immune expression have been fairly well elaborated.
During the COVID-19 crisis, melatonin has primarily been lauded for its anti-inflamatory effects, though there is ample evidence that it has just the opposite effect in immunocompromised individuals. In fact, it is commonly recommended specifically for immune enhancement at orally supplemented doses many times higher than natural secretion rates. Such doses have been shown to powerfully enhance cytokine production, thereby improving immunodeficiency.523
While there has been conflicting evidence on melatonin’s actions in various immunological contexts, it has been repeatedly demonstrated to provoke autoimmune disease in susceptible individuals.524
Therapeutic benefits
Before we dive into an examination of melatonin’s dangers in the context of viral infection, let’s first take a look at its beneficial therapeutic effects.
Melatonin is a systemic antioxidant
Melatonin essentially has two modes of action: 1) activation of its receptors followed by downstream signaling cascades and 2) direct actions as an electron donor to specific free radicals. This is an important distinction for our discussion. It is specifically its antioxidant actions which are thought to be non-localized and systemic.525526
Melatonin possesses antiviral activity
Regarding antiviral activity, melatonin does increase the production of interferons along with up-regulated IL2 expression. The latter specifically enhances myelopoiesis leading to an increase in granulocytes, monocytes, macrophages, and dendritic cells. This is a particularly important consideration, especially in the context of higher viral loads, when granulocyte activity is out of control, with parallel lymphopenia.527
Melatonin stabilizes cell membranes
Beyond antioxidant activity, melatonin has also demonstrated the ability to stabilize cell membranes, thereby making them more resistant to oxidative damage.528 In fact, it will even enter the nucleus and protect damage to DNA directly, so in that sense, it is working cellularly “from the inside out” under elevated oxidative stress.529530
Melatonin inhibits chemokines
Melatonin can, in certain contexts, inhibit chemokine expression, thereby reducing neutrophil and macrophage/monocyte infiltration. However, studies demonstrating this ability have consistently failed to consider the entire immunological landscape, focusing primarily on pro-inflammatory mechanisms, such as NF-kappaB. It has been claimed that melatonin inhibits transcriptional regulation of chemokines such as CCL2 by directly blocking PI3K/Akt-induced NF-kappaB and downstream STAT-GAS signaling pathways, but disease status and local immune processes in tissues/organs are vitally important for interpretation.
In fact, in the context of chronic viral infections, melatonin, at physiological doses, is actually accompanied by increased lymphocyte proliferation and inhibition of IL10, a context in which chemokine activity would be expected to increase. As mentioned above, melatonin up-regulates IL2, which increases chemokine responsiveness and higher recruitment of antigen-activated T cells into sites of infection/inflammation. The question arises: where is the “switch” that determines whether melatonin promotes myelopoiesis or myelosuppression?531532
When we consider studies demonstrating melatonin’s ability to block infiltration of neutrophils and macrophages, we must deeply understand the full picture and the context within which it is operating, as will become more evident below as we discuss melatonin’s dangers.533
Melatonin reduces adenosine A2b receptor expression
Adenosine receptors are most frequently associated with xanthines such as caffeine, which antagonize them, producing stimulating effects of varying degrees. Adenosine is released in response to multiple stimuli including hypoxia, tissue damage, and chronic inflammation, which agonizes adenosine receptors throughout the body. In the lungs, in fact, adenosine A2b receptor activation causes bronchoconstriction.534
Further, all receptor subtypes (A1, A2a, A2b, and A3) are expressed on a wide variety of cells, including immune cells. A2a and A2b, in particular, are recognized as mediators of inflammation.535536
Another way in which melatonin modulates inflammation is by reducing A2b receptor expression, specifically.537
Dangers of Melatonin in COVID-19
If we take a closer look at the specific interactions of melatonin with the hematopoietic system which produces myeloid and lymphoid cells, it becomes more clear that its effects can vary widely depending on multiple immunological factors.
GM-CSF provokes melatonin’s dark side
One of the contexts in which melatonin can become decidedly pro-inflammatory is during the excessive neutrophil and macrophage infiltration into the lungs that characterizes severe ARDS in COVID-19. The up-regulation of these cell types depends on the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF), which provokes stem cells to produce granulocytes and monocytes. Monocytes then mature into macrophages and dendritic cells, thereby initiating the inflammatory cascade. Macrophages, in turn, secrete GM-CSF, further increasing their numbers. For this reason, high GM-CSF levels are seen in bronchoalveolar lavage fluid at later stages of this disease.
In this environment, melatonin can be problematic for a number of reasons. First, melatonin directly activates CD3+/CD4+ T helper cells, increasing IL2 levels. At the same time, melatonin enhances IL12 production by monocytes, driving T-cell differentiation towards a Th1 phenotype — this up-regulates antiviral IFN-gamma. On the surface, this sounds like a desirable effect — from the perspective of antiviral immunity.538
However, at physiological levels in serum, melatonin will synergistically work with IL2 to further activate peripheral T-cells, increasing expression of its own receptors, systemically.539
Activated T-helper cells will, in turn, produce more GM-CSF, leading to overall increased immune expression. This may be of therapeutic benefit in immunocompromised individuals, but at later stages of the disease or in those prone to immune hyperactivity (i.e. autoimmune disorder), it could make inflammatory potential decidedly worse.
This is precisely why immune status is of vital importance in determining the applicability of melatonin for COVID-19 therapy. In the presence of hematopoietic suppression, increasing GM-CSF levels to promote granulocyte and monocyte differentiation is a highly desirable outcome. These individuals are more prone to complications from viral or bacterial infections, given the lack of innate and adaptive immune responses.540
In such individuals, low doses of the opioid antagonist Naltrexone has proven effective in promoting increased beta-endorphin secretion — a very important factor in the consideration on melatonin.541
You see, beta-endorphin activation of mu-opioid receptors is an important part of antiviral antibody production, though they have shown a decidedly biphasic effect. When beta-endorphin levels go too high, antibody production is substantially reduced. Lower concentrations, however, actually stimulate antiviral antibodies. Therefore, immunocompromised individuals are often also low endogenous opioid producers, resulting in insufficient beta-endorphin concentration to allow antigen-driven specific antibody production. Low-dose Naltrexone is frequently used in such contexts to boost humoral immunity. What is interesting about beta-endorphins is that they have the exact opposite effect on natural killer cells. Higher levels increase NK-cell concentrations. It is for this reason that we often see paradoxical effects in strategies that use low-dose Naltrexone (LDN) for immunological support. We often hear conflicting reports from doctors saying LDN can both stimulate immunity and improve outcomes in autoimmunity. This seems contradictory until we consider the biphasic nature of the opioid system along with individual potential for beta-endorphin secretion and pre-existing immunological status.542
I am taking you off on this tangent with Naltrexone for a reason: activated T-helper cells from both lymph nodes and bone marrow may be stimulated by melatonin to release very specialized opioids called “melatonin-induced opioids” or MIOs, similar in activity to endogenous opioids. As you might expect, they have the same biphasic effects on immune expression. This brings us to an important consideration: MIOs and their relationship to antibody production.
With regards to antibodies, it would appear that melatonin may have the direct ability to block Fc receptor expression. FCGR2B, for example — an immunoglobulin receptor responsible for the phagocytosis of immune complexes and regulation of antibody production by B-cells — is down-regulated by high-dose melatonin treatment. This might be of considerable benefit for individuals with higher viral loads who are at risk for antibody-mediated inflammation. For those with impaired humoral immunity, however, such an effect could be quite dangerous.543
MIOs, also knows as opioid cytokines, bind to both mu and kappa opioid receptors, similar to endogenous beta-endorphin and dynorphin, respectively. Mu-binding will either up-regulate antiviral antibody response at low ligand concentrations, whereas too much — and we will see humoral suppression. Likewise, high MIO activity at mu-receptors can also increase natural killer cell response, which is very desirable for acute antiviral response, but provides no longer-term protection. Kappa receptor binding, in contrast, effectively shifts macrophages from an M1 inflammatory phenotype to an M2 anti-inflammatory one.544545
Physiological doses of melatonin in immunocompromised individuals can provoke T-helper response and up-regulate immune protection. At increasing doses, however, there will be greater MIO-mediated suppression of antibody response, thereby increasing the risk for prolonged replication or worse, re-infection after recovery. The question is: what will the effect be in a COVID-19 patient with skewed immune response or those at higher risk for autoimmunity? You can probably guess the answer by now.
Physiological doses of melatonin in individuals with normal or only mildly “flagging” immune response will promote increased concentrations of GM-CFU which, in turn, amplifies macrophage activity. If given chronically, this strategy could modulate immunity in unfavorable ways.546
It should not be surprising that the immunoenhancing effects of melatonin can be completely neutralized with Naltrexone, indicating it is, in large part, mediated by interactions with the opioid system. What does this say about its relationship to lung injury, inflammasomes, and acute respiratory distress?547
Melatonin can invoke autoimmunity
The truth of the matter is that autoimmune reactivity is a very real risk with physiological melatonin doses, and I do not see that those recommending its liberal use in clinical contexts have sufficiently addressed this concern. It is not enough to make vague proclamations that “everything has two sides”. We must quantify each side and understand their variability in proper context.548
Consider that activated T-cells are significantly more responsive to melatonin and show heightened binding activity with it.549 Increased binding to membrane or nuclear melatonin receptors in T-helper cells/monocytes stimulates the secretion of cytokine opioids (MIOs) along with interferon-gamma, IL2, IL1, IL6, and IL12.550
Indeed, nocturnal peaks in melatonin secretion have been associated with higher Th1/Th2 ratios — e.g. high IFN-gamma and low IL10.551 In fact, nocturnal spikes in melatonin production have been correlated with elevated IL12 and nitric oxide production by macrophages in patients with autoimmune rheumatoid arthritis. When melatonin is injected in the synovial fluid of such patients, it also shows increased levels of IL12 and nitric oxide.552
Ironically, the same mechanism in melatonin that provokes autoimmune reactions is precisely how it provides protection against viral encephalitis and bacterial infections — namely, via amplified myelopoiesis.553
To reiterate, this is a desirable early-stage response to reduce viral loads, but as discussed above, increasing melatonin doses will produce ever-increasing GM-CSF fueled neutrophil/macrophage activity, leading to granulocyte accumulation and inflammation. Nowhere else is this pattern more evident than in cases of pediatric tonsillar hypertrophy, which are accompanied by high levels of melatonin. And where else do we see granulocyte accumulation? — Rheumatoid granulomas.554555
Obviously, it should be of paramount importance to the clinician where on the immunological spectrum a patient is if considering physiological doses of melatonin, especially if there is any pre-existing history of autoimmunity or familial predisposition to it.556 Apparently, dose matters, and as I have mentioned earlier, higher doses would be expected to exacerbate MIO-mediated disruption of antibody response, regardless of immune status.
Unfortunately, there is a twist that involves sex: if you are a male, your chances of melatonin-induced autoimmunity and related inflammation-driven injury are significantly higher. It has been well established in recent years that androgens are protective against autoimmune disorders, whereas estrogens can provoke them. In fact, autoimmune diseases are notably more severe in females than in males, and in animal models, females experience earlier autoimmune-correlated mortality.
The reasons for this disparity are somewhat counterintuitive — females show overall higher levels of total IgG, which correlated with more robust humoral (i.e. antibody-mediated) immunity.557558
It should not surprise you at this point that the administration of melatonin in females reduces their humoral response, thereby decreasing total IgG antibodies and reducing inflammatory potential. In male subjects, however, the effect is actually the complete opposite.559
As we have firmly established, enhanced activation of macrophages leads to increased inflammation with elevations in TNF-alpha, IL1B, IL6, and nitric oxide — all of which play a role in tissue damage.560561
Disparities in melatonin response between men and women, however, basically comes down to antiviral interferon-gamma: melatonin decreases high IFN-gamma in females and increases low IFN-gamma in males. IFN-gamma, in turn, promotes the production of IgG2a and IgG3, enhancing antibody-mediated hyper-inflammation.562
This is a critical problem for those with autoimmunity, in that such shifts in IFN-gamma mediated IgG provokes MHC expression and autoantigen presentation to what would otherwise be sleeping non-tolerant, anti-self T-cells. That is, IFN-gamma in this context is waking up autoreactive T-cells, promoting local immune reactions and inflammatory cascades.563564
Immunosuppressive IL10 levels are also varied by sex, as they are normally lower in females and increased by melatonin, whereas melatonin lowers potentially IL10 expression in males.565
To clarify this further: females are predominantly high in IFN-gamma and low in IL10, whereas males (due to androgen activity) have a predominantly anti-inflammatory profile with lower IFN-gamma, higher IL10 but, ironically, greater susceptibility to viral infection. Melatonin appears to shift these dynamics, moreoever, in a hormone-specific way.
In fact, melatonin suppresses estrogen production, thereby protecting females from an otherwise pro-inflammatory immune response with autoantibodies.566567568
Testosterone, on the other hand, has been proven to have just the opposite action on the immune system.569570571
Therefore, as a potential treatment in COVID-19, melatonin has a decidedly amplifying effect on pre-existing autoimmunity and, in those so susceptible, could actually promote such disease. This risk is significantly higher for men than for women. To complicate matters further, hormone fluctuations such as estrogen dominance in men or peri-menopause in women can modulate effects as well. Where are you on this spectrum?
The summary of the issues is as follows: for individuals at risk for auto-immune disorder, melatonin may be beneficial for younger females and risky for younger men. The lower your estrogen level, the less likely melatonin will provoke an autoimmune response. Therefore, prior to engaging in therapies that might include this hormone, hormone status and autoimmune markers such as TGF-beta and T-regulatory cell levels may be helpful.
Dangers of melatonin in daylight hours
Given that melatonin is a circadian hormone, its synchronization with immunological processes is time-sensitive. For example, low melatonin levels at night have proven to enhance the development and growth of tumors. In fact, melatonin administration late afternoon or in the evening has inhibited tumorigenesis in a variety of clinical models.
Some studies, however, have described melatonin’s capability of actually promoting tumor growth if taken during the daylight hours. This evidence would significantly reduce the treatment window, especially in people with low natural-killer cell counts — such as those with long-term, chronic viral infections (e.g. Epstein-Barr). A proper dosing threshold for NK-cell enhancement prior to therapy would be mandatory, given that lower doses can diminish such counts.572573
Verdict: Melatonin should be used with great caution
The takeaway for melatonin is that while it may be a powerful antioxidant, anti-inflammatory, and immunomodulator, it should not be embraced with complete confidence without first knowing, at the very least, current immune status, history of autoimmunity, hormone levels, and — most importantly — stage of disease.574
It should further be noted that the pineal gland is located outside the blood-brain barrier, and some reports have indicated that IFN-gamma may actually modulate melatonin secretion there, in response to viral infections.575 Keeping this in mind, it seems that the purported “immunoenhancing” effects of melatonin appear to be restricted to T-dependent antigens and would be more pronounced in individuals with immunosuppression.
Such immune-deficient states could, ultimately, be equated with age, since reduced hematopoietic stem-cell renewal and balanced differentiation are gradually lost with time. But in younger, healthier individuals or those with a predisposition to autoimmune disorder, melatonin is clearly a much greater risk. Double that risk if you are a young woman or man with uncontrolled estrogen levels.576577
Nonetheless, we continue to see announcements (mostly coming from China) that melatonin receptor agonists greatly reduce lung edema, pro-inflammatory cytokines levels in bronchoalveolar lavage fluid, and apoptosis in lung tissue resulting from the use of ventilators. One such study published in March, 2020 has been cited endlessly across social media, however, no one seems to have acknowledged that the test subject was a rat.578
Other studies further claim that melatonin directly blocks activation of the NLRP3 inflammasome, but again — the majority of the inflammasome studies are using models of sepsis or LPS-induced lung injury which present very different immune profiles than would be seen in SARS-CoV-2 infection — further, the majority of those studies were also using rat subjects.579
To be brutally honest, evidence of successful reversal of ARDS or ventilator-induced lung injury in COVID-19 patients is extraordinarily sparse, and for good reason — melatonin has not been adopted unanimously by global health professionals as a COVID-19 treatment nor has it demonstrated widely applicable efficacy. And the reason why should be abundantly clear: it has biphasic effects that depend explicitly on the immunological status of the patient and their degree of viral load.
Therefore, I highly advise clinicians to approach this strategy with great caution and do their own in-depth research. All too often, the proponents of single-compound strategies side-step evidence that could place their therapeutic “cure-all” in a negative light. But this is not insomnia, chronic inflammation, or other such condition for which melatonin could be marginally helpful. Individuals that should know better are promoting its ability to prevent SARS-CoV-2 infection and reverse ARDS, and such claims, unfortunately, are extremely irresponsible and ultimately dangerous. As a global community, we should do our best to filter such information sources, given their appeal to innocent bystanders looking for quick, efficient over-the-counter protections.
 Ascorbic Acid: Overhyped and Dangerous Redox Molecule
Ascorbic Acid: Overhyped and Dangerous Redox Molecule
Another such example of “molecular hagiography” is the curious but unfortunately passionate Vitamin C movement, which lately has been lauding ascorbic acid as the ultimate cure for COVID-19 — and any other disease you might have, for that matter. Ascorbic acid offers a unique opportunity for would-be researchers and self-proclaimed experts to leverage decades of well-established confirmation bias in service to “revolutionary” and “ground-breaking” advances in nutraceutical science.
How Linus Pauling Started a Runaway Train
If we were to point a finger at anyone for starting this endless carousel of pseudoscience, the blame would have to go to Dr. Linus Pauling. For Pauling, what could have been an unblemished, exemplary career in science, culminating in a 1954 Nobel Prize for the discovery of alpha-helix protein structures — ended with a long, protracted and painful parade, promoting vitamin C as the “cure for cancer” — a disease he (and his wife) succumbed to in 1981 and 1994, respectively — regardless of the endless crates of ascorbic acid powder they had tunneled through on their way to the grave.
In truth, ascorbic acid deserves a very special place in the category for irresponsible and reckless therapies recommended for the resolution of COVID-19. With each decade that has passed since the publication of Pauling’s first book “Vitamin C and the Common Cold” in 1976,580 ambitious researchers have piggy-backed on one partial truth after another, building a large “information tree” of increasingly abundant low-hanging fruit for those that would continue to carry the torch.
And therein lies the problem: the majority of studies surrounding ascorbic acid, its properties, and its clinical applications are categorically one-sided. It is very rare that you will find a study that examines this molecule from a negative angle (of which there are many), and even more rare that one of its researchers is open to debate about his / her findings. The reason for this phenomenon is simple — scientists with a more comprehensive education and prestigious reputation are not inclined to touch vitamin C research with a 10-foot pole. Just the mention of this molecule in a scientific paper can (and has) ruined careers, for nothing more than the pseudoscientific history it carries with it. Unfortunately, such limitations have led to an increased number of eager fringe researchers, looking to make their reputation with “the universal redox molecule”.
This aggressive and territorial trend for ascorbic acid research was, in fact, first exhibited by Linus Pauling himself, when, in 1978, his talented research assistant, Arthur Robinson, Ph.D., released a report that megadoses of vitamin C (i.e. 5-10 grams per day) could actually promote some types of cancer in mice.
In fact, according to Robinson’s account, nearly all of the mice developed skin cancers (squamous cell carcinomas) following exposure to ultraviolet radiation. A total of 1,846 hairless mice received 38 different diets, and the rate of onset and severity of tumors varied by as much as 20-fold.
On average, the animals being fed ascorbic acid according to Pauling’s recommendations contracted skin cancer nearly twice as frequently as the control group. Ironically, it was only the nearly lethal doses that had any protective effect.
Pauling responded to this report quite resolutely — he forced Robinson to resign from the institute, slaughtered all of the experimental animals, impounded all of Robinson’s data, destroyed all previous research results, then committed libel, saying Robinson’s research was “amateurish and inadequate”. Robinson sued the institute, Pauling, and its trustees, and the dispute was eventually settled out of court for $575,000. At today’s rates, that’s about $2.3 million. And thus began the legacy of “vitamin C can do no harm”.581
Since that time, ascorbic acid has not performed all that well under scrutiny. At least 16 well designed, double-blind studies showed that vitamin C does not prevent colds. With regards to cancer, three early studies demonstrated ascorbic acid was no better than placebo, and countless other well-designed, randomized, controlled trials (in pill form) confirmed such findings.582583
Those studies that did prove anti-cancer effects were, in general, full of uncertainties, including the use of retrospective controls, complete lack of independent pathologic confirmation, and no blinding or placebo use.584585586587
Nonetheless, this molecule continues to attract its proponents, each one committing the same error one after the other, approaching research with a confirmation bias and selectively ignoring contradictory evidence.
More recently, the use of intravenous sodium ascorbate has demonstrated slightly different effects than regular oral form, and this has provoked renewed interest in the “magical powers of the molecule”. In the months since the onset of the COVID-19 crisis, it has been promoted as having the ability to prevent or even reverse acute respiratory distress syndrome (ARDS) — a conclusion rooted in, unfortunately, a lack of basic knowledge in molecular biology, virology and, as it turns out — hemodynamics.588
Let’s dissect the premises, one concept at a time, in order to clarify why using ascorbic acid for COVID-19 could well be the most reckless strategy of all. But first, I would be doing a disservice by not reviewing the potential therapeutic benefits.
Therapeutic benefits — with a caveat
During critical illness, plasma and cellular levels of ascorbic acid are rapidly consumed, along with other antioxidants such as glutathione, primarily to neutralize inflammation-mediated oxidation events.
The majority of studies on this phenomenon have focused on patients with sepsis, reperfusion, and burns, so very little may be said about antioxidant status in COVID-19. We can only presume, due to the massive cytokine storm it can provoke, that antioxidant depletion will be a problem.589
Nonetheless, we are seeing recommendations now for massive doses of vitamin C, mostly in IV sodium ascorbate form, based primarily on these sepsis studies along with sparse clinical evidence that only vaguely tracks all the biomarkers that would be required for concrete, unbiased conclusions.590
Given ascorbic acid’s solubility in water, it is found as a cofactor in a large number of metabolic processes. Though it can be prooxidant in a narrow band of contexts (which I will describe below), it’s primary activity is as an antioxidant, donating electrons for the neutralization/scavenging of free radicals. Ascorbic acid also prevents the generation of new free radicals — via NADPH oxidase (NOX) pathway suppression — and supports the recycling of other antioxidants. In this respect (and perhaps the only valid one), ascorbic acid can relieve oxidative burdens systemically. Unfortunately, with higher doses, its behavior is far less predictable, depending on a wide array of factors.591
Ascorbic acid has very broad influences over immune expression, and the contextual nuances often evade researchers. With regards to SARS-CoV-2, where there is frequent derangement of neutrophil to lymphocyte ratio, ascorbic acid will have decidedly paradoxical effects. On the one hand, it may conditionally promote the proliferation of T cells and natural killer cells, having a supportive effect on antiviral potential. Ascorbic acid will also enhance neutrophil chemotaxis and phagocytosis which, under lower viral loads, can support clearance. On the other hand, as the disease progresses, neutrophil infiltration in airway epithelial cells is actually one of the triggers for pathogenic inflammation, and any “enhancement” of chemotaxis can only be problematic.592593594
Further, it appears ascorbic acid may induce antibody production in B cells which, for some individuals, as I have described above, can also exacerbate inflammation.595
In models of animal sepsis, administration of ascorbic acid improves outcomes by inhibition of T-regulatory cell activity. To be clear, this is a beneficial outcome in the context of immunosuppression and lymphopenia, but could provoke negative outcomes in individuals susceptible to autoimmune disorder and excess T-cell mediated inflammation — incidentally, a condition that is more likely to occur at earlier stages of COVID-19 progression.596
With regards to macrophages, it would appear that vitamin C deficiency, at least in sepsis models, leads to lower macrophage activity and impaired migration. Again, improving macrophage expression in this scenario is therapeutic, but not in hyper-inflammatory states with macrophage accumulation — another common ARDS biomarker. I want to make it very clear here that discrepancies between ascorbic acid’s antioxidant and immune-stimulating activities are not very well delineated in existing studies. Presumptions are often made either by referring to in vitro results elsewhere or by drawing correlations between the presence of ascorbic acid (in vivo) and changes in immune response. I don’t need to remind you that oxidative stress and immune gene up-regulation go hand-in-hand, it is often difficult to place the “chicken before the egg” in such scenarios.
If anything, it should be stressed that ascorbic acid-induced neutrophil chemotaxis is a highly oxidative process, and we cannot presume 1-to-1 relationships between immune stimulation and anti-oxidant homeostasis.597
Nonetheless, it would appear that the anti-oxidant potential of ascorbic acid, specifically, may be capable of actually blocking T-cell activation. You see, concrete evidence of ascorbic acid-mediated enhancement of T-cell proliferation has been primarily based on petri dish cultures. This is an important distinction when we consider that extracellular vitamin C in serum is most often in its reduced form (ascorbate), whereas in vitro, it is instantaneously oxidized to DHA (dehydroascorbate).598
In other words, vitamin C uptake in vivo and in vitro are likely to differ greatly, and petri dish results cannot be used to determine what is actually happening in peripheral blood lymphocytes. In reality, these contexts are substantially different in character.
For example, peripheral blood lymphocytes (PBLs) in vivo are known to accumulate up to 80-fold higher concentrations of ascorbate than in serum, particularly in the elderly, and this has been confirmed in studies showing high lymphocyte ascorbate levels. Nonetheless, at increasing doses, the intracellular DHA to ascorbate ratio increases in lymphocytes. Why does this happen?599
The answer may surprise you — as mitochondria-derived reactive oxygen species (ROS) levels accumulate in T-cells, lymphocytes are gradually activated, acquiring “oxidative burst potential”, an effect that peaks about 1-2 hours after activation. As a matter of fact, other investigators demonstrated antioxidants / free radical scavengers, applied during T-cell activation, can thoroughly shut down IL2 secretion and, thereby, stop proliferation.600601602603
In other words, ascorbate does not activate T-cells at all — quite the contrary — their activation is oxidatively mediated by dehydroascorbate, which enters the cell via glucose transporters. Once inside, intracellular DHA is reduced back to ascorbate at the expense of glutathione (GSH), provoking increased “oxidative burst” and further inducing the T-cell to secrete IL2 leading to proliferation.
This, in turn, creates a chain-reaction called the “bystander effect”. Activated cells release more superoxide, promoting the oxidation of extracellular ascorbate to DHA which is then rapidly taken up by neighboring cells and reduced to ascorbate intracellularly (commonly known as the “recycling” mechanism) — again, producing oxidative burst in those cells at the expense of GSH. The in vitro experiments show T cells internalizing more petri-dish oxidized DHA when they are activated, due to enhanced glucose transporter (GLUT1 and GLUT3) expression that persists up to 48h after activation. Make no mistake about it — this will only happen in vivo if there is sufficient DHA outside the cell. In fact, blocking the oxidation of ascorbic acid to DHA in vitro almost completely inhibits uptake.604605 It stands to reason that extremely high doses of ascorbate may have unexpected results, especially in those individuals that are deficient in glutathione.
Megadoses such as those proposed for COVID-19 could, therefore, suppress T-cell proliferation, IL2 secretion, and CD25 expression by reducing intracellular ROS levels. In fact, such an effect was achieved in one in vitro study by adding just 0.5 micromoles of vitamin C 30 minutes prior to activation.606
Therefore, we have yet one more example of a disconnect in experiment design among the ascorbic acid proponents. It would seem the only way that ascorbate could promote T-cell activation and proliferation in vivo would be in a highly oxidized environment extracellularly, where ascorbate is rapidly being oxidized to DHA. And when you consider that T-cells reside in the periphery before homing into infected sites, it would seem necessary for there to be excess pre-existing (systemic) oxidative stress for the conditions to be proper for T-cell activation. Otherwise, ascorbate will enter such cells through the sodium-dependent SVCT2 transporter, neutralize ROS, and pre-empt oxidative burst before it’s had a chance to occur. Are the clinical trials using ascorbate looking at the neutrophil to lymphocyte ratio (NLR) before and after initiation of treatment? You can easily guess the answer to that question.
Ascorbic acid has paradoxical effects on the cardiovascular system as well. The positive side is that it decreases lipid peroxidation, mostly via antioxidant effects in the vasculature, and improves blood flow by directly inhibiting TNF-induced intercellular adhesion, resulting in the lower leukocyte adhesion to the vascular walls.607608609610611
Furthermore, it would seem that ascorbic acid also has the capability of decreasing binding affinity for angiotensin II to the AT1 receptor in vascular smooth muscle cells. Nonetheless, there is absolutely no evidence that would support the use of vitamin C for any significant blood pressure lowering effects. Quite the contrary, in fact, high-dose ascorbic acid is frequently used in patients with septic shock to increase blood pressure. Its primary mechanisms in this context include nitric oxide synthase inhibition, improved vascular responsiveness to vasopressors, increased norepinephrine and cortisol synthesis in the adrenal medulla, and improvements in vascular endothelium integrity.612613
In fact, ascorbic acid plays a vital role in both the catecholamine and vasopressin synthesis pathways. For catecholamines, it serves as a cofactor for the copper-containing enzyme, dopamine β-hydroxylase, which synthesizes norepinephrine from dopamine. Ascorbic acid deficiency has actually shown to decrease norepinephrine levels, especially in the adrenal glands systemically, thereby lowering blood pressure. Ascorbic acid is also essential for the generation of pituitary hormone vasopressin which increases fluid retention also for the purpose of increasing blood pressure.614615
The problem here is two-fold: late-stage COVID-19 patients with higher ACE2 down-regulation and Angiotensin II levels would be much more vulnerable to complications resulting from enhancement of hypertension and AngII directly stimulates both norepinephrine and vasopressin secretion. With high-dose ascorbic acid infusions in this environment, there would be higher potential for exacerbating vasopressor effects, even considering ascorbic acid’s secondary AngII inhibiting activity. Further, though BH4 is technically a cofactor for nitric oxide production as well, it is isoform-indiscriminate and, in cytokine storms, will predominately participate in reactions for inducible NOS, elevating levels of nitrosative stress, systemically. As I will describe in more detail below, this is a very real problem in models of hemolysis with haptoglobin suppression, given ascorbic acid’s ability to reduce methemoglobin to oxyhemoglobin which, in turn, aggressively scavenges intravascular nitric oxide.616
Clear and present dangers
Ironically, in the process of describing therapeutic benefits for ascorbic acid, we can’t but help step on its toes due to the unexpected effects that might be seen, depending on a mind-boggling number of factors including antioxidant, immunological, and cardiovascular status. I can assure you that a comprehensive analysis of all of these factors is not being performed prior to injecting massive doses of ascorbic acid into patients. As with Dr. Pauling and every researcher that built on his research with this molecule — positive results are isolated, amplified, and reported, while any potentially contradictory evidence is often systemically suppressed. For this reason, you don’t see a lot of double-blind, placebo-controlled, randomized studies demonstrating ascorbic acid’s clinical efficacy. In reality, if we investigate this molecule without confirmation bias and a critical eye for paradoxical metabolic reactions, a much darker picture emerges.
G6PD deficiency: Severe Consequences
The most severe complication of all with regards to high-dose ascorbic acid therapies, is the presence of glucose-6-phosphate dehydrogenase deficiency (G6PDD). G6PD is an enzyme that participates in the pentose phosphate pathway — supplying reducing energy to cells, especially red blood cells — by maintaining levels of NADPH. This NADPH in turn maintains proper levels of glutathione, helping protect the red blood cells against oxidative damage. Individuals with G6PDD are therefore unable to properly regenerate glutathione and are extremely vulnerable to oxidative stress, due to drops in NADPH levels. In fact, even the process of hemoglobin recycling between oxygenated and deoxygenated forms will generate oxygen radicals inside RBCs.
In most cases, G6PDD is not noticeable, due to backup dehydrogenase reactions that produce NADPH. Under excess stress, however (such as during viral infection), NADPH production starts to lag and reduced glutathione (GSH) levels sharply decline. As I have described above, ascorbic acid may be oxidized by cells producing oxygen radicals, but it may also participate in ROS generation on its own by reducing metals such as copper and iron, an effect I will describe in greater detail below.
Human RBCs rely on glucose transporters and are preferential for DHA absorption. In fact, they are deficient in the ascorbate binding SVCT2 transporter, and so rapidly accumulate DHA, requiring reduction back to ascorbate by dehydroascorbate reductase (DHAR), using glutathione. In an environment with excessive oxidative stress, NADPH is being chronically oxidized for the production of superoxide. This leaves little NADPH behind for use by glutathione reductase — which reduces oxidized glutathione (GSSG) back to GSH. The end result is GSSG and DHA accumulation, uncontainable intra-RBC oxidative stress, and eventually, hemolysis.617618619620
Since the 1980s, members of the ascorbate contingent have been trying to prove the persistence of an ancient cytochrome membrane oxidoreductase as a potential recycling mechanism for extracellular monodehydroascorbate. It was long ago established that this cytochrome (Cytb561) arose in the course of evolution to reduce extracellular iron during erythroblast differentiation. Nonetheless, stubborn pseudoscientists have clung to the theory that it might play a major role in ascorbate recycling. A study published in 2006, for example, asserted this function (again — in vitro), yet declared that “proof of the cytochrome’s MDHA recycling capability is not yet available but could be provided by differential expression studies”.621
Not surprisingly, a group of Chinese scientists took up the challenge to prove this cytochrome’s expression in 2014 by performing “in silico” electron transfer modeling of the Cytb561 protein structure, extracted from Arabidopsis thaliana, a flowering plant (chosen either for its vague homology to human proteins or its ubiquitous presence in the cracks of sidewalks in suburban China). This study — highly reminiscent of the computer models that recently supported far-fetched theories of SARS-CoV-2’s attack on hemoglobin — allegedly “proved” the capability of Cytb561 to effectively re-cycle extracellular MDHA to ascorbate in transmembrane fashion — without the slightest hint of NADPH/glutathione. Keep in mind they came to this conclusion using nothing more than a flower’s DNA expressed in E. Coli culture.
That being said, the Cytb561-mediated electron-transfer, proposed by Lu et. al.622 — if it were true — would present problems for an RBC in an oxidative environment. Intracellular ascorbate would pass an electron across the membrane via Cytb561 to extracellular MDHA, leaving MDHA behind in the cytosol. In the process, ferric iron would also be reduced to ferrous iron. The problem with this model is that MDHA is, itself, a radical and if not rapidly reduced, will disproportionate into ascorbate and DHA, the latter of which would further accumulate in oxidative environments. The extracellular ferrous iron, now in the Cytb561’s extracellular “pocket” would be free to oxidize in other reactions. It should be obvious, even with this proposed transmembrane re-cycling mechanism, that these dynamics would not play out well during rampant viral infection.
To come to the point: to date, no one has proven (or ever will prove) that a conserved transmembrane cytochrome capable of saving RBCs from oxidative burden exists. This is yet one more truth-stressed hypothesis coming out of the fantastical world of “vitamin C biology” whereas, in clinical contexts of hemolytic stress, no mechanism has surfaced to save these cells apart from improving NADPH/glutathione status. It hasn’t stopped ascorbate proponents, however, from citing such studies as fact.
To reiterate, systemic oxidative stress will increase both plasma and cellular DHA significantly, especially at sites of infection. High-dose ascorbate, therefore, increases DHA levels considerably in such contexts. Normally, RBCs have a high capacity for recycling of DHA back to AA, but this capacity is lost — either in circumstances where there is systemic NADPH/glutathione depletion or worse, G6PDD.623624
Glutathione levels plummet in severe infections or any other model of rampant oxidative stress, along with available NADPH, and as this cascade progresses, RBCs with or without G6PD deficiency, will lose their ability to recycle dehydroascorbate, eventually leading to oxidatively-mediated hemolysis. In fact, this is one of the ways in which COVID-19 increases permeability of lung vasculature — not by the mythological “ORF-protein hemoglobin attack complex”.
Ascorbic acid liberates iron
Next, there is the question of granulocyte uptake of DHA and its effects on iron metabolism. This is a topic that has not been well covered, yet it has extraordinarily troubling implications.
During SARS-CoV-2 infection, widespread oxidative burst in granulocytes produces a highly oxidative environment, especially at locations of viral activity. As should be quite clear by now, this increased local oxidation will rapidly “consume” any and all available ascorbate, leaving it in its oxidized DHA form.
Granulocytes, similar to T-cells, accumulate DHA through up-regulation of their GLUT transporters, further increasing their potential for oxidative burst. The rest, as they say, is history — the “bystander effect” takes hold, extracellular ascorbate is systemically reduced to DHA, and oxidative stress amplifies. The more ascorbate you feed into this system, the more potential for ROS there will be. It is not a well-behaved “antioxidant” in this environment. It is being hijacked by granulocytes for excessive antiviral oxidative activities.625
To make matters worse, free-iron, which is sequestered into ferritin in an inflammatory environment, will be resolutely released from ferritin in the presence of ascorbate.626 This effect has been primarily studied in the context of cancer, though it may be seen anywhere that iron-binding proteins encounter a low pH.627
In addition to direct ascorbate-mediated iron liberation from ferritin, the resulting oxidized dehydroascorbate will be taken up by granulocytes, thereby stimulated to release superoxide, liberating even more ferritin iron.628
For those cells that have higher sodium-dependent SVCT2 expression, accumulation of ascorbate can actually reduce iron or copper ions, keeping them available for highly oxidative Fenton reactions. In this way, high-dose ascorbate can induce mass lipid peroxidation directly via its interactions with iron.629
Once iron has been released, it could potentially sustain further hydroxyl radical production. This is troubling when you consider that granulocytes contain small amounts of ferritin and that superoxide dismutase neutralization of superoxide generates hydrogen peroxide, which will in turn react with free iron to amplify Fenton reaction potential. Remember how the presence of higher levels of hydrogen peroxide has been noted in the breath of asthmatics? How will they fare with increased ascorbate-mediated release of ferritin iron and granulocyte oxidative burst?
We have a dual problem here: extracellular DHA may be taken up by granulocytes to promote oxidative burst while accumulating re-cycled ascorbate may simultaneously reduce excess oxidation and prevent apoptosis, prolonging cell life.
Therefore, when you hear an ascorbic acid proponent lauding its potential to improve neutrophil chemotaxis and phagocytosis, consider the total environment and its potential to provoke granulocytes to excessive, pathogenic behavior. In this environment, mega doses of antioxidants can become clearly pro-oxidant. We can presume glutathione status would have much more profound implications in such circumstances.
Ascorbic acid deranges hemodynamics
We have already touched upon how this molecule might nudge iron in the wrong direction, but there are further details that can make it much more dangerous in the presence of pre-existing hemolysis.
Iron complexes have a highly variable reactivity. Free ferrous iron, which is present in cell-free oxyhemoglobin, reacts with both nitric oxide and hydrogen peroxide, forming nitrate and hydroxyl radicals, respectively. Regardless of its fate, ferrous iron will either strongly scavenge nitric oxide or produce potently oxidative radicals via a Fenton reaction. Only in its ferric form, however, will it bind to transferrin.
These reactions are amplified with either increased iron absorption and uptake (via diet or supplementation) or via iron release from cell-free hemoglobin or ferritin. Both of these scenarios are influenced by ascorbic acid. On the one hand, ascorbic acid increases dietary iron absorption. In circulation, it further reduces ferric iron to ferrous form, where it can become more reactive with NO and hydrogen peroxide, and directly promotes the release of iron from ferritin, especially during oxidative stress.630
One would presume, on the surface, that reducing ferric-iron containing cell-free hemoglobin (i.e. methemoglobin) back to ferrous oxyhemoglobin is a desirable strategy — and in fact, such strategies have been deployed when administering transfusions in order to maintain hemoglobin’s ability to bind oxygen. Nonetheless, I must remind you that these approaches only work when there is adequate endogenous production of haptoglobin and / or hemopexin, both of which are consumed at higher rates during hemolysis.
If mega-doses of ascorbate are administered in this environment, it will strongly increase nitric oxide scavenging and hydroxyl radical production by oxyhemoglobin. Haptoglobin levels must parallel oxyhemoglobin, and its capacity may be easily outstripped with increasingly higher levels of ascorbate.631632
During SARS-Cov-2 infection, elevations in hydrogen peroxide and neutrophILreleased hypochlorous acid will push oxyhemoglobin to such reactions, which is why we frequently see suppressed or non-existent haptoglobin levels in sepsis.633
To further clarify: cell-free hemoglobin arises as a result of either increased oxidative stress in RBCs (from loss of glutathione for ascorbate recycling) or from mechanical pressure in contexts of vascular permeability, both of which cause deformation or damage to the erythrocyte and release of hemoglobin into circulation and/or extravascularly. Oxyhemoglobin will quickly react with nitric oxide intravascularly and, at sites of inflammation/infection, directly with hydrogen peroxide.
Ascorbate will not be able to help in either circumstance if, prior to hemolysis, glutathione has been depleted or, post-hemolysis, ascorbate has already been oxidized to DHA by granulocyte oxidative burst. Administering ascorbate for methemoglobin reduction to oxyhemoglobin is only relevant in transfusions / HBOC administration when there is little to no pre-existing oxidative stress.
What I am trying my best to drive home here is simple — COVID-19 is characterized by increased chemotaxis of neutrophils and macrophages (CCL2 / IL8 mediated), elevated pro-inflammatory cytokines, and massive oxidative stress — all of which will favor oxidation of ascorbate to DHA, resulting in less and less ability for reductive recycling.
How will megadoses play out in variations of this scenario with different degrees of antioxidant deficiency and or immune dysregulation? In my opinion, we cannot say. And unfortunately, the clinical trials with ascorbate all suffer from chronically impaired design and follow-up, so we have little more than vaguely improved mortality rates to gauge outcomes.
That being said, COVID-19 has not been shown to be a disease of profound hemodynamic derangement. High free-iron levels have not been reported in serum, nor have we seen excessive cell-free hemoglobin either in blood draws or in bronchoalveolar lavage fluid. Moderate hemolysis is commonly seen in acute respiratory distress, so this is nothing new for COVID-19.
There is a serious question of just how relevant cell-free hemoglobin or iron metabolism is, ultimately, and whether the results seen with ascorbic acid administration to COVID-19 patients may be characterized by any other mechanism than the obvious — reduction of overall oxidative stress, similar to melatonin.
Such as it is, with iron overload or hemolytic diseases, ascorbic acid deficiency is protective, given lower potential for tissue iron mobilization. In fact, higher doses of ascorbic acid are used precisely with this purpose in mind — to draw iron out of tissues and bind to chelating agents in hemoglobin disorders with iron overload.634635
To further round out this discussion of iron metabolism, it should be noted that the potential for iron load could be magnified in COVID-19 in situations where there is even mild hemolytic release of cell-free hemoglobin in combination with a history of higher iron consumption. Further, ascorbic acid increases iron’s absorption and can raise body iron levels over time, especially in men and postmenopausal women.
Dissolved intestinal ferrous or ferric form iron is readily absorbed by the duodenal mucosa, but such absorption will be further enhanced by either ascorbic acid or diets with sources of citric or lactic acids (the latter of which is abundant in fermented foods). Further, diets high in phytates, such as found in grains, legumes, and nuts will have the opposite, inhibiting effect — and given that paleo / ketogenic diets are becoming more popular these days, such foods are likely to be eliminated, changing iron absorption dynamics. Therefore, there are many ways in which excess iron could become a problem. Ascorbate would significantly complicate that playing field.636637638
For example, in addition to provoking the release of iron from ferritin, ascorbate may also locally lower pH, thereby inducing release of iron from transferrin as well.639 Further, ferrous iron formed by reduction of ferric iron by ascorbate will not bind to transferrin. Ferric form iron is required for transferrin binding and sequestration. Therefore, ascorbate will promote higher free ferrous iron until it is converted to ferric via reactions with oxygen radicals. For all of these reasons, if there is any net free iron accumulation of any kind during therapies using megadoses of vitamin C, the consequences are quite serious, resulting in widespread oxidative damage, such as that seen in hemochromatosis.640
Even with moderate elevations in free iron, possibly as a result of enhanced absorption in the digestive tract, ascorbate, along with other reducing agents, will contribute to iron-initiated damage by catalyzing the reduction of ferric iron(III) to ferrous iron (II), which in turn carries out systemic free radical formation wherever it is found.
And one last note on the topic of iron metabolism: melatonin has also been shown to possess reducing power for ferric iron, dose-dependently. Therefore, therapeutic strategies combining ascorbate and melatonin will be expected to increase the potential for ferrous-iron / oxyhemoglobin reactions with nitric oxide and hydrogen peroxide, as stated above.641
High-dose ascorbic acid hastens kidney damage
Hopefully, it should be abundantly obvious by now that ascorbic acid has a great many hidden sides that few are talking about due to a lack of appreciation for the variations in clinical context. One more such unfortunate area is the kidneys.
With potential for RAS dysregulation, hypertension, and vascular permeability, the kidneys will be first in line for oxidative insult. And administration of ascorbate in the context of any degree of kidney disorder will lead to acute free radical generation locally.
In those individuals with high serum ferritin, as little as 300mg injected ascorbate can interact with trapped iron in ferritin and potently invoke iron release.642
Nonetheless, we frequently see nephrologists recommending 50 to 200mg ascorbate doses for hemodialysis patients, three times a week. Such doses have conditionally proven to sidestep resistance to EPO (erythropoietin) in individuals with iron deficiency.643644
That being said, the elephant in the room here is ferritin, and iron storage status is rarely checked in these situations.
Ferritin makes or breaks ascorbate’s clinical potential in models of kidney disease, and with increasing ferritin levels, the potential for increased oxidative damage elevates exponentially. In fact, hemodialysis patients are known to be highly susceptible to abnormal oxidant production and are lacking in basic antioxidant protections.645
There have been numerous reports over the past two decades of increased lipid peroxidation in hemodialysis patients receiving intravenous iron infusion.646 As we have already discussed, ascorbate will be highly pro-oxidant in the presence of free iron. To reiterate, ascorbate can mobilize iron from inert tissue stores, tightly correlating its oxidative potential to inert tissue stores rather than total free iron.
In addition to oxidative damage in the kidneys resulting from ascorbate-mediated iron liberation, there is also the additional risk of nephropathy from oxalate accumulation. Multiple reports demonstrating acute oxalate accumulation with high-dose ascorbate have surfaced over the years. In every case, there was some degree of pre-existing renal insufficiency or stress.647
Potential for oxalate accumulation may be increased in hemolytic disorders, given cell-free heme’s toxicity in the kidneys, especially in hemopexin deficient states.648 The risk for this problem seems to be increased with oral administration. Case in point: intestinal absorption of ascorbate is rate-limited, so it could remain for long durations in the alkaline environment of the small and large intestines, favoring oxidation to oxalic acid. Due to sodium-transporter mediated absorption, it would end up in circulation as sodium oxalate.649
Circulatory half-life of oxalic acid is just over 3.6 hours, prolonging the excretion rate to 14 hours.650 It has been surmised, 6 hours after ingesting 100g of ascorbic acid, there will be roughly 80mg of oxalic acid excreted, just under the 100mg oxalic acid / day seen in individuals with kidney stones.651
The “bright” side of this consideration is that calcium oxalate stones take months to years to develop, and this generally only happens when excretion rates are from 100-400mg per day.652
Nonetheless, more rapid accumulation and resulting complications are possible in direct correlation to individual glomerular filtration rate. The lower this rate, the more potential for ascorbic acid and oxalic acid accumulation. For this reason, mega-doses of ascorbic acid are a real risk for individuals with pre-existing kidney disease or stress, exponentially speeding up time to stone onset.
Further, this rate limitation applies only to oral doses of 500mg / day or more. Massive intravenous doses of sodium ascorbate could easily bypass this barrier many times over.653654
While intravenous ascorbic acid is less prone to oxidative reactions with oxalic acid than high-dose oral form, such solutions may nonetheless lose stability over time, so freshness of substrate and expediency would be determining factors for potential kidney damage.655656657
There is another side to the oxalate inquiry, however, that makes it far more pertinent in the context of COVID-19. It involves the way in which oxalate metabolism shifts during respiratory disease, in particular. A perfect example would be asthma and COPD, both of which have biomarkers that, as we have discussed, dovetail with SARS-CoV-2 provoked acute respiratory distress.
Even mild serum elevations in oxalate can have serious implications when there is any degree of vascular permeability in the lungs. Oxalate, in fact, may be detected in bronchoalveolar lavage fluid, and in higher amounts can provoke “respiratory oxalosis”. Therefore, while oxalate damage to the kidney may take longer time to inflict damage, small fluctuations in serum oxalate during respiratory distress would rapidly increase alveolar damage. Here, it is important to emphasize that oxalate is not only an end-point for ascorbic acid metabolism but also a component of many foods that, alone, could increase serum oxalic acid levels.
Such foods include spinach, rhubarb, almonds, cashews, beets, cocoa powder, raspberries, stevia, sweet potatoes, and more. High intake of such foods in parallel with high-dose ascorbic acid, for any duration, further amplifies oxalate load — something that may not be visible for many months or even years but could quickly and fiercely present itself during acute respiratory distress, especially in contexts of vascular permeability and hemolysis.
Once again, the studies surrounding this effect do not track the necessary biomarkers nor observe intake over a sufficient duration of time. Therefore, there are conflicting opinions regarding ascorbic acid’s oxalate-increasing potential. Nonetheless, the presence of oxalate in bronchoalveolar lavage fluid should be concerning, especially with comorbid kidney disease and / or high-dose vitamin C. In this respect, intravenous infusions could promote the most severe damage, under the “right” circumstances.658
Calcium oxalate and iron accumulation in sarcoidosis
Another relevant angle on the oxalate issue involves macrophages. Sarcoid granulomas are known to sequester significant amounts of iron, stimulating ferritin production and giant cell formation.
In vitro studies have demonstrated calcium oxalate’s ability to foster granuloma formation by increasing the release of IL8 and IL6. In vivo, these macrophage clusters contain crystalline oxalate which interacts with iron on the crystal’s surface. In the case of excessive iron release from ferritin by, for example, ascorbic acid, there is a higher potential for dysregulation in iron-oxalate interactions leading to further granuloma formation.
One study actually demonstrated increased pulmonary iron and ferritin accumulation after intratracheal instillation of calcium oxalate crystals.659 The increased iron, in turn, is accompanied by an increased chemotactic (i.e. IL8 mediated) influx of macrophages.
Even without granuloma formation, it is clear that in conditions that increase serum calcium, oxalic acid, or free iron — in combination with higher intake of ascorbic acid — can amplify macrophage and neutrophil chemotaxis, provoke release of iron from tissue ferritin, and thereby prolong exposures to immune-associated oxidative stress. Consider that such scenarios could be fatal with chronic mega-dosing which would gradually lower glutathione status, deplete recycling capability, and elevate oxidized dehydroascorbate systemically via the mechanisms I have described above.
Ascorbic acid enhances intracellular DNA damage
In this inflammatory milieu, ascorbic acid has yet another unpredictable behavior.
During cytokine storms, there is rapid, unrelenting depletion of most bodily antioxidants, including vitamin C. Depending on the window of time in which mega-doses of vitamin C are administered, this can either reduce oxidant stress or, quite the opposite, powerfully promote it. The latter outcome would be expected in individuals that are already ascorbate-depleted and are suddenly infused with a large bolus of, for example, sodium ascorbate.
Once again, we have the dilemma of comparing the results of in vitro studies with in vivo clinical practice. Such comparisons inevitably lead to contradictions. For example, in vitro, ascorbate is capable of scavenging peroxynitrite — a free radical formed from the reaction between superoxide and nitric oxide.660
Peroxynitrite is particularly dangerous in that it reacts with both protein and non-protein thiol residues, inhibits mitochondrial respiration, promotes lipid peroxidation, incurs DNA damage, and eventually triggers mitochondrial permeability transition and apoptosis.661662663664665
As it turns out, cells with significantly increased levels of ascorbic acid are far more susceptible to the DNA damaging effects of peroxynitrite.
Uptake experiments performed on U937 cell lines — which, as I have mentioned earlier, behave similarly to mature macrophages — consistently reveal large ascorbate accumulation. This is, in part, due to dehydroascorbate-mediated oxidative burst, perpetual recycling, and “bystander effects”, as described above. It is precisely for this reason we see high ascorbate accumulation in patients’ white blood cells during infusions.666
Here, it is important to be reminded that as ascorbate is oxidized to DHA, both superoxide and hydrogen peroxide are formed.667 In locations with higher nitric oxide concentrations, such as the vasculature or at sites of infection, there is concomitantly higher potential for reaction with hydrogen peroxide derived from ascorbate’s oxidation. In fact, the extent of potential DNA damage in the presence of 100 micromoles of peroxynitrite is far less than that seen with only 20 micromoles of peroxynitrite together with ascorbate.
Therefore, during cytokine storms, large doses of ascorbic acid / sodium ascorbate, as proposed by the vitamin C proponents, could magnify the potential for peroxynitrite formation exponentially, thereby inducing widespread DNA damage. It is critically important to note here that this damage could very well be entirely “silent” during the duration of treatment and many months or, more likely, years post-treatment. Cancers and tumors are slow-forming processes, which is why adverse oncogenic reactions to ascorbate are difficult to quantify. Linus Pauling’s cancer is the perfect example and even he was praising the molecule for “delaying cancer onset for decades”, a claim that could never be scientifically verified.
Studies indicating that ascorbate is cytoprotective fail to appreciate that at increasingly higher doses, the potential for oxidation to DHA increases, thereby elevating hydrogen peroxide volume, systemically. Ironically, it is specifically this hydrogen peroxide elevating capability that is quoted as therapeutic in the context of cancer.668
And let’s not forget that macrophages accumulate iron via their CD163-receptor-mediated scavenging of hemoglobin-haptoglobin (Hb-Hp) complexes — a problem that is amplified in conditions with hemolysis.
Rapid rises in macrophage ascorbate levels would quickly reduce transition metals (e.g. iron) and mobilize them from intracellular stores, thereby increasing peroxynitrite-dependent damage. In this way, high-dose ascorbate may induce widespread oxidative damage. In fact, this is precisely what we see in models of idiopathic hemochromatosis and dietary iron overload.669670671
Further, ascorbate powerfully enhances susceptibility of monocytes and macrophages to peroxynitrite toxicity, raising the potential for DNA damage to bystander cells — a situation that would be unavoidable during runaway inflammatory immune responses.
Ascorbic acid can induce vascular stress
Under more normal conditions, where chronic immune-mediated oxidative stress is not an issue, ascorbic acid will have the opposite effect by accelerating the degradation of S-nitrosoglutathione and thereby increasing nitric oxide, lowering blood pressure.
Further, in such normal conditions, vitamin C would be expected to normalize vascular hyper-responsiveness to norepinephrine, perhaps in part by reducing binding affinity of angiotensin II to AT1 receptors. In such circumstances, there is less activation of NADPH-oxidase by AngII receptor binding and, thereby, lower vascular sensitivity to catecholamines.
For the reasons I have stated above, mega-doses of vitamin C could behave quite differently if there were pre-existing derangements of such systems, as seen in SARS-CoV-2 infections, particularly at later stages.672
The primary risk, however, from my perspective, is in ascorbic acid’s ability to reduce cell-free methemoglobin to cell-free oxyhemoglobin, which powerfully scavenges nitric oxide, thereby potentially elevating blood pressure. This effect would not be prominent in individuals with sufficient haptoglobin but, again, haptoglobin deficiency is common in models of hemolysis. The question remains — just how common is such hemolysis in COVID-19? To re-iterate, we haven’t seen indicators of iron overload in patients thus far. Nonetheless, individuals prone to such overload are at considerably higher risk, especially when mega-dosing ascorbate, given its ability to mobilize iron from ferritin.
Verdict: Ascorbic acid is unpredictable and should be avoided
Last October (2019), a randomized controlled trial was carried out, dubbed the “CITRIS-ALI RCT”. It involved the administration of about 15g / day IV ascorbic acid for 4 days in 167 patients with sepsis-related ARDS. Similar to other ascorbic acid studies, the conclusion was that vitamin C, compared with placebo, did not significantly improve organ dysfunction scores or alter makers of inflammation and vascular injury. Nonetheless, the call for “further investigations” is made. History repeats itself.673
This was not the first trial to look at vitamin C’s use in clinical contexts. Another trial combined vitamin C, hydrocortisone, and thiamine to treat patients with pneumonia, again with less than compelling results.674675
Not surprisingly, a brand new placebo-controlled trial in Wuhan, China was announced in April, 2020 investigating vitamin C infusion for the treatment of severe SARS-CoV-2 infected pneumonia. It proposed the use of IV sodium ascorbate to treat 140 patients at a dose of 24g / day for 7 days. While this trial is expected to come to completion in September, 2020, we can already predict vaguely positive results surrounded by great paucity of essential quantitative biomarkers.
While we cannot peer into the trial’s design, the conclusion may be made, nonetheless, that there will be the glaring absence of documentation for iron status, pre-existing kidney / cardiovascular / respiratory disease, medications, lifestyle/diet, and all the other numerous factors I have listed above. Further, we can also presume that whatever the result of the trial, the vitamin C contingent will ignore all “contradictory noise” in the data, embrace any degree of success (regardless of similarity to placebo), and then resolutely reference the trial as proof that ascorbic acid may be used liberally for not only the treatment of COVID-19 but all other viruses that stem from its family into the future.
Here’s how to make it work
If you are a patient or clinician that is hell-bent on applying this strategy, regardless of the warnings I have provided above, it would be useful to re-iterate critical conditions that must be met in order for it to be successful:
- Categorically avoid ascorbic acid if there is any degree of kidney disease, whatsoever. Even minor increases in oxalate can increase the risk of tissue damage and resulting declines in glomerular filtration.
- Similarly, patients with any degree of G6PD deficiency should also avoid high-dose ascorbate. Consider parallel infusion with glutathione to maintain proper ascorbate recycling and avoid oxidative stress. As stated above, transmembrane ascorbate recycling mechanisms are purely theoretical and have not been proven in vivo. Clinical experience would refute their existence, in general, as glutathione depletion strongly correlates with ascorbate-induced intracellular stress.
- It should be a pre-requisite for all strategies using high-dose ascorbate to either verify haptoglobin status and/or administer recombinant haptoglobin in parallel. This will prevent ascorbate-mediated oxyhemoglobin reactions with nitric oxide and hydrogen peroxide, as described above.
- Ascorbic acid should likely be avoided in patients with already high neutrophil:lymphocyte (NLR) ratios, as its promotion of T-cell differentiation is highly oxidative. Further, ascorbate’s potential to increase neutrophil chemotaxis in the context of ARDS is not desirable, as it could exacerbate lung inflammation.
- Increased doses of thiamine can improve glyoxalate aminotransferase-mediated degradation of glyoxalate to carbon dioxide instead of oxalate. Higher ascorbic acid doses increase the potential for oxalate accumulation, especially in alveolar macrophages of the lung, and thiamine could mitigate this outcome.676
- Avoid ascorbic acid in the presence of chronic hemolysis or iron overload, as it can exponentially amplify oxidative stress and resulting tissue damage.
Even with keeping all of these conditions under strict control, I would strongly advise the medical community at large to hold ascorbate-centric research with utmost skepticism and avoid rushing to conclusions about universal applicability. Its behavior appears to be very far from predictable, and the variance in results correlates tightly with both a patient’s pre-existing condition and current immunological status.
The best advice for this molecule would be to do your own research and categorically avoid opinions that are exclusively one-sided and / or combined with “dosing instructions”, the latter of which is highly irresponsible and dangerous, regardless of whether it is connected with a credentialed medical professional or not. Make no mistake about it: wild variations in individual genetics and clinical contexts can produce paradoxical results with any “dual” redox molecules. Cancer and tumor pathologies take time to develop — and just one brief, high-dose encounter with ascorbate could induce enough low-level damage to evolve into problems only 10-years later or more.
Please do your best to not be emboldened by the ascorbate studies showing DNA-protective potential at guanine bases, while minimizing or ignoring other types of mutagenic damage. Any damage is an oncogenic risk — both a blazing forest fire and a small match can, ultimately, bring down a house. Ascorbic acid puts out a fire with one hand while inciting one with the other. And based on decades of research, it is impossible to say which side of that equation a patient will land, especially after prolonged, high-dose use. Therefore, let us all do what we can to move on and finally accept that vitamin C neither cures colds nor cancer — and most certainly not COVID-19.
 Vitamin D
Vitamin D
The majority of people in modern times are vitamin D deficient, mostly due to increasingly sedentary lifestyles combined with lower sun exposures. I will leave a discussion of UV-induced melanoma risk for another article. With regards wot COVID-19, it should be strongly emphasized that adequate 25-hydroxyvitamin D levels are protective, though levels of 70 ng/dL or more may be increasingly risky, primarily due to effects on calcium balance.
The correlation between vitamin D levels — specifically active-form calcitriol — and sarcoidosis (i.e. acute macrophage accumulation) was proposed well over 50 years ago. At that time, serum elevations in calcium were noted. As we have already discussed, macrophages up-regulate their own expression of 1-alpha hydroxylase to increase calcitriol levels. In fact, one of the ways in which corticosteroids help in sarcoidosis is by suppressing 1-alpha hydroxylase expression in macrophages.
While sarcoidosis has not been explicitly observed in COVID-19, many of its immunological features are similar — especially excess macrophage chemotaxis and accumulation at infection sites. Further, at least in the early stages, we also see activation and proliferation of mature T-cells secreting cytokines such as IL2 and IFN-gamma, both of which have repeatedly been observed in sarcoidosis.677
Therefore, it is no stretch of the imagination to presume that alveolar macrophages in SARS-CoV-2 infections also produce higher levels of calcitriol. So far, I haven’t seen any specific measurements of 1,25-hydroxyvitamin D, but it may be advised to better quantify vitamin D status and its effects on immune expression in this context.678
To further clarify, most calcitriol is derived by converting circulating 25-hydroxyvitamin via 1-alpha hydroxylase in the kidneys. It can also be synthesized locally by macrophages expressing the same enzyme, so the rate-limiting factor is— 25-hydroxyvitamin D, which is the most commonly supplemented form, also known as D3.
It is quite possible that one of the reasons lymphocyte counts decline in COVID-19 is due to macrophage-derived calcitriol’s ability to down-regulate their expression, via inhibition of IL2 and IFN-gamma.679680
This inhibition is, in fact, the body’s attempt to down-regulate T-cell activity at sites of inflammation. Unfortunately, with increasing levels of 25-hydroxyvitamin D, this suppression could become excessive.
It is interesting to note that hydroxychloroquine has also been shown to inhibit 1-alpha hydroxylase and lower calcitriol levels, a property that may not be desirable in those that are vitamin D deficient.681
That being said, calcitriol measurements could be an indirect way to gauge macrophage activity and, thereby, immune status in COVID-19 patients. It should be noted that calcitriol elevations are also correlated with autoimmune conditions, and it can only be presumed that this is a regulatory mechanism to suppress overactive immune response.682683684
As 25-hydroxyvitamin D levels rise, there is an elevated potential for calcitriol-mediated increases in serum calcium. This can lead to higher calcium excretion in urine, thereby amplifying the potential for calcium oxalate stones, especially if there is parallel high-dose intake of ascorbic acid.685
Therefore, both dietary calcium and vitamin D3 status should be quantified prior to implementing any therapeutic strategy in COVID-19. There is ample evidence of high D3 levels in sarcoidosis, but even without frank presentation of this disease, simple alveolar macrophage accumulation and heightened chemotaxis could produce similar clinical outcomes.686687
Selenium
There have been some concerns expressed regarding the use of selenium/selenomethionine during SARS-CoV-2 infection due to reports of selenocompounds having ACE-inhibiting capability.688 Whether or not ordinary daily intake of this mineral presents a problem has not been established. In fact, if selenium — in doses up to the recommended daily allowance — were to have notable ACE-inhibiting capability, it would have shown potential to improve outcomes for patients with hypertension (it hasn’t).
The truth of the matter is that selenium is absolutely essential for containment of oxidative stress and has decidedly antiviral effects.689690 In fact, selenium deficiency has been correlated with genomic mutation in viral RNA and increased potential for virulence.691
I will, nonetheless, play devil’s advocate and consider the outcome — if selenium, indeed, could substantially inhibit ACE. My perspective on such concerns is similar to that of the other ACE inhibitors and angiotensin-receptor blockers (ARBs) we have already discussed.
On the one hand, ACE inhibition can increase ACE2 expression, thereby providing a wider “attack surface” for CoV-2 virions On the other hand, ACE2 shedding induced by spike-protein binding is a far greater concern. To re-iterate, with loss of ACE2, while there is less potential for viral entry, there is also a greatly lessened ability of ACE2 to convert AngII to the vasodilatory, cardioprotective septapeptide Ang(1-7). So with increasing loss of ACE2, there is an exponentially greater risk for hypertension and vascular remodeling / permeability — along with the serious consequences that flow from that.
Selenium would be expected to confer the same benefits as ARBs, which I have exhaustively elaborated earlier in this article. At the same time, it would also provide both effective antiviral support and thyroid protection, by support of glutathione peroxidase activity. Therefore, it is my opinion that concerns surrounding selenium’s use during COVID-19 are just as unfounded and those regarding ACE inhibitors and ARBs.
Preventing lung damage is of just as high a priority as reducing viral replication. Both must be embraced together. Selenium avoidance for the purpose of reducing ACE2 expression is, essentially, an ill-conceived premise that does not fully consider the broader dynamics of RAS-system modulation in the context of COVID-19. Therefore, the idea that selenium could be contraindicated for SARS-CoV-2 is, unfortunately, another unfounded conclusion coming out of the same group of self-proclaimed experts that promote melatonin and vitamin C. One can inflict exponentially greater damage with those nutraceuticals than with this all-essential mineral.692
 BiopathFx: Customized Therapeutics
BiopathFx: Customized Therapeutics
At Transcend Genomics, we utilize advanced genome-based protein modeling, transcriptomics, and interactome studies to better understand how diet, lifestyle, and supplementation play into genetic expression. Rather than relying exclusively on genome-wide association studies, as the majority of other genetics-based services do, we instead attempt to determine individual variability outside the restraints of limited-population statistics.
In layman’s terms, we don’t believe there is a “one-size-fits-all” strategy for COVID-19 and, in order for therapies to be effective, they must take into consideration all of the factors laid out in this article, at both the genetic level and through comprehensive analysis of laboratory biomarkers.
Using our BiopathFx methodology, we have analyzed all of the genes correlated with SARS-CoV-2 infection and progression and modeled each relevant pathway against known pharmaceutical and natural interventions. The result is a more comprehensive, potentially more effective, list of strategies that could be applied, in a modular fashion, to patients in order to best serve their individual needs with the least amount of potential clinical complications.
To be very clear, such strategies will not work without properly gauging what stage of the disease a person is in. As should be abundantly clear from the concepts shared in this article, what may be supportive in early stages could trigger complications later on, when inflammation levels are rising and there is less control over immune regulation.
I would like to share with you a small selection of curated compounds that our BiopathFx methodology has categorized as both therapeutic and “anti-therapeutic”. This will hopefully give you a perspective on how we are currently deriving prophylactic strategies for clients. These are not to be interpreted as silver bullets nor even applicable to the public at large. Rather, they are a starting point for your own explorations and research. Again, I want to emphasize that specific recommendations, doses, and other such specifics should never be shared publicly in any medium or context, and those that do so clearly cross lines of morality and integrity. Only by knowing yourself and quantifying your own weaknesses will you be able to guide your own path to health, ideally with the help of a qualified and open-minded medical professional.
Therapeutic Candidates
Resveratrol
This natural phenol is produced by several plants in response to pathogen-induced injury. While it is more well known for its beneficial effects on cardiovascular disease, cancer, metabolism, and (controversially) lifespan, it actually has some very interesting properties that may make it a powerful ally in the fight against COVID-19.
-
Decreases neutrophil and macrophage chemotaxis693694
-
Improves glutathione-conjugation of toxins in liver695
-
Modulates haptoglobin production696
-
Inhibits NLRP3 inflammsome and ameliorates lung injury697698699
-
Promotes mitochondrial biogenesis700701702
-
Directly lowers the expression of TMPRSS2703
Zinc
Zinc is absolutely vital for proper immunity and deficiency is common in tuberculosis, pneumonia, acute lower respiratory tract infection, and the common cold.704
-
Inhibits replication potential in coronaviruses705
-
Useful in the early stages of the disease by directly improving interferon-mediated antiviral immunity706
-
If taken during cytokine storm, it will suppress IL10, amplify chemotaxis, and increase inflammatory potential, so care must be taken with timing.707
Quercetin
This flavonoid is most abundantly found in citrus fruit, buckwheat, and onions. It is mostly known for its ability to inhibit allergic-type inflammation, though antioxidant and protein kinase enzyme inhibition activities have also been noted. Before implementing this compound, it should be noted that it is decidedly estrogenic, with binding potential for both alpha and beta estrogen receptors. Nonetheless, other potential attributes have been documented that could prove useful in COVID-19.
-
Reduces angiotensinogen expression, thereby preventing elevations in angiotensin II708
-
Suppresses excessive inflammation709
-
Protects against vascular injury / remodeling710
Simvastatin
Beyond lipid-lowering effects, this statin also has a wide range of potentially protective effects throughout the course of coronavirus infection.
-
Ameliorates angiotensin-II induced endothelial dysfunction711
-
Decreases excessive granulocyte chemotaxis712
-
May decrease viral loads and mitigate inflammation713
-
Has shown the ability to inhibit the NLRP3 inflammasome714
Low-Dose Dipyridamole (LDD)
Dipyridamole is a nucleoside transport inhibitor and a PDE3 inhibitor medication that inhibits blood clot formation and prevents stroke when given chronically, long-term. Short-term use, however, promotes blood vessel dilation and lowers blood pressure.
Lately, low-dose dipyridamole (LDD) has been proposed for the treatment of insomnia, dry eye, dry mouth, erectile dysfunction, and fibromyalgia. Most notably, however, is its ability to prevent and treat viral diseases in both adults and children. Though very little is known regarding its off-label mechanisms, much may be presumed about its molecular activity, given it is an analogue of adenosine.715
-
Modulates lung epithelial inflammation via adenosine receptor A2a716
-
Augments IL10 response717
-
Protects mitochondrial membranes against iron-induced lipoperoxidation718
-
Decreases metalloproteinase expression and release by monocytes719
Metformin
Originally marketed as an anti-diabetic drug, Metformin is now gaining traction for its anti-aging potential. While it would be beyond the scope of this article to explain why Metformin may actually be a dangerous option, long-term, for either blood glucose control or attainment of longevity, it nonetheless possesses some very useful properties in the context of COVID-19.
-
Reduces chemokine expression720
-
Inhibits intravascular inflammation721
-
May improve antiviral protection by up-regulating type I interferon signaling722
-
Increases mitochondrial biogenesis723
-
Inhibits NLRP3 inflammasome724
N-Acetyl-Cysteine (NAC)
Acetylcysteine is an N-acetyl derivative of the amino acid L-cysteine. L-cysteine, along with glycine and glutamine, is an essential precursor for the formation of master-antioxidant glutathione. I have already described the potential in COVID-19 for massive depletion of all antioxidants systemically. Glutathione plays a particularly important role in red-blood-cell antioxidant capacity, without which there is a greater risk for hemolysis.
-
Inhibits ACE (angiotensin converting enzyme)725
-
Reduces AngII receptor binding726
-
Reduces macrophage hyperactivity and promotes dormancy727
-
Reduces vascular inflammation by inhibiting AGEs / glycation728
-
Improves parameters of COPD729
-
Reduces excess mucous in the lungs, though this may be problematic for those with genetics for lower mucin production.730
Compounds to Avoid during COVID-19
Acetaminophen
Marketed as Tylenol, this non-steroidal anti-inflammatory drug’s safety for SARS-CoV-2 infection has not been thoroughly proven. Nonetheless, the medical community at large has mostly focused on ibuprofen’s risks and recommended, with impunity, the use of acetaminophen for pain and inflammation reduction. Let’s look at various ways this drug could be problematic for COVID-19.
-
Decreases intracellular glutathione in pulmonary macrophages and type II pneumocytes, resulting in profoundly diminished antioxidant capability in the lungs731
-
Depletes glutathione in the liver, potentially leading to hepatic necrosis, especially in the presence of pre-existing, systemic oxidative stress732
-
May increase systolic and diastolic blood pressure in patients with pre-existing cardiovascular disease733
Dexamethasone
The use of corticosteroid medications has been mostly discouraged for COVID-19 patients, due to their potential to suppress immune response. Obviously, timing is everything — suppressing immune response at the early stages of infection can inhibit antiviral potential whereas at later stages when immune processes are out of control (e.g. during cytokine storms), a certain amount of suppression might be desirable. That being said, there are other mechanisms of action that make corticosteroids such as dexamethasone problematic for this disease.
-
Increases gene expression of angiotensin II type 1 receptors734
-
Induces ACE expression in monocytes, paradoxically increasing their inflammatory potential735
-
Can alter T-cell expression, amplify chemokine response, and trigger autoimmunity, especially in the elderly736
-
Substantially decreases glutathione in alveolar epithelial cells, enhancing potential for oxidative damage737
Copper
We have already discussed the dangers of excessive serum iron and how that might be exacerbated by high-dose ascorbic acid intake. Copper, however, also plays a significant role in disease outcome. Not only does it compete for transluminal transport with iron and zinc, but it can, at increasing levels, also suppress zinc’s immune-benefits.
-
Copper is required for inflammasome activation, and chelation blocks NLRP3 induction738
-
Excess copper transport in pulmonary arteries contributes to the development of hypoxia-induced pulmonary hypertension.739
-
Ascorbic acid “auto-oxidizes” in the presence of copper, in a dose-dependent manner740
This is not to mean that copper should be avoided altogether. On the contrary, a proper zinc to copper ratio (generally from 15-30:1 zinc:copper) fosters not only robust immunity but also balanced expression of superoxide neutralizing SOD2, protecting mitochondria from oxidative stress. As copper levels rise in the direction of (or higher than) zinc, hydrogen peroxide from superoxide dismutation can further react with free copper to form hydroxyl radicals — a reaction that would be promoted by ascorbic acid. Therefore, the admonition is to keep zinc and copper in the right ratio and avoid large doses of ascorbic acid which have already been described above to be decidedly unpredictable and potentially dangerous.
DHEA
This naturally occurring adrenal steroid hormone is a metabolic intermediate in the production of both testosterone and estrogen. Its popularity has risen over the years, especially among longevity proponents because its levels are known to decline with age. While its potential benefits (in those that are deficient) are numerous, there may be several reasons why it may need to be avoided during SARS-CoV-2 infection.
-
May induce T-cell proliferation and increase natural killer cell count, but these effects are only seen at higher doses after at least 12 weeks time.741
-
Inhibits antiviral interferon-gamma and induces expression of IL10 and TGF-beta, both of which are immuno-suppressive. This effect occurs about 1 month into supplementation.742
-
Protects against vascular inflammation, pulmonary hypertension, and lung remodeling / fibrosis.743
-
Improves nitric oxide levels and increases vaso-relaxation744
-
Directly opposes corticosteroid effects, potentially promoting TNF-alpha induced inflammation745
Overall, the immune-modulating effects of DHEA appear to have a delayed onset, but may be in effect for people that have already been supplementing with it prior to infection. Nonetheless, it could inhibit antiviral immunity and promote inflammation (through corticosteroid opposition) at the wrong times, so using it in a clinical context would be challenging, to say the least.
Nicotine
It might surprise you to see nicotine in the list, but apparently, it has been recommended (in chewing gum form) by certain fringe health groups, so a brief discussion of its dangers is in order here.
-
The most obvious problem is nicotine’s ability to transiently but nonetheless powerfully stimulate vasoconstriction, and thereby elevate blood pressure.746
-
Nicotine enhances ACE2 expression directly in the lung (via inhaled products) but more systemically if taken sublingually or via patch.747
-
Induces pro-inflammatory potential in alveolar macophages748
-
Enhances adhesion potential in endothelial cells, promoting macrophage-mediated inflammatory damage749
-
Provokes cathepsin-dependent activation of NLRP3 inflammasome750
-
Activates dendritic cells and augments their capacity to stimulate T-cell proliferation and cytokine secretion751
Synthroid / Excess T4
Last but certainly not least, we have the thyroid hormone T4, administered in either synthetic form (Synthroid) or as part of naturally desiccated thyroid products (NDT). The problem with these therapies is that in individuals that have slow or impaired conversion of T4 to the active form, T3, there is the potential for T4 pooling and higher conversion to reverse T3. This results in an overall down-regulation of metabolic rate — something that is already happening during infection. To make matters worse, excess T4 can also have other undesirable effects.
-
Thyroxine (T4) up-regulates angiotensinogen, thereby promoting higher AngII and increasing blood pressure.752
-
Directly induces the expression of pro-inflammatory cytokines such as IL1B and TNF-alpha753
-
Provokes superoxide production in alveolar neutrophils and macrophages754
Nonetheless, proper levels of thyroxine, in balance with T3, are vitally important for immune balance. Studies have shown thyroxine’s ability to potentiate interferon-gamma mediated antiviral activity, so if you suffer from hypothyroidism and are taking medication that contains T4 / thyroxine, the advice is simply to ensure you are not in excess. This can be confirmed by measuring free-T4/free-T3 ratio along with reverse T3.
 COVID-20: What’s Coming Next?
COVID-20: What’s Coming Next?
With each day, our knowledge continues to evolve and we grow closer to developing sustainable, effective therapeutic strategies for this pandemic. Depending on when you are reading this article, the COVID-19 storm may either be waning or already passed us by, but regardless — this is not the end of our complicated relationship with the world of virions. Quite the contrary, what we are dealing with in hospitals and clinics now is not the same viral genome we encountered at the onset of the original crisis.
Significant antigenic shift and drift have already been observed across the coronavirus family. SARS-CoV-2, further, has demonstrated rapid rates of mutation and adaptation. Unfortunately, this does not bode well for those that would put all of their bets on a CoV-2 targeted vaccine. Clearly, this family of viruses, and likely many other families as well that we have not yet become acquainted with, are actively adapting and widening their attack surface in multiple organisms outside of Homo sapiens species.
As with the epidemics that came before COVID-19, we weren’t aware of the presence of these viruses until they hopped into our phylogenetic branch of the tree. Therefore, it can be said with considerable certainty that viruses are actively adapting, in part provoked by environmental stressors and other such inputs, and will be expected to become more virulent with time. That is, their ability to infect host cells will improve. If we shut down one receptor or block it’s binding now, it will eventually adapt and find entry by some other means. We need to outsmart our adversary, and we need to act fast.
Therefore, it is absolutely paramount that we develop modular strategies that are not specific to one viral strain or even this family but can allow us to also expedite adaptation and workarounds for viral mutations effectively.
This pandemic has brought out a lot of obtuse, emotionally charged opinions about life, death, and mortality. Some have been saying that viruses are a planetary regulatory mechanism for limiting overpopulation of specific species. While it is far beyond the scope of this article to discuss population dynamics and the idea of “carrying capacity”, suffice it to say that our species has done quite a bit of damage to the ecological hierarchy within which we exist.
Obviously, it is pure speculation to presume that viral activity could be working against us in order to maintain ecological balance — almost as useless as the idea that this virus has been developed for the purpose of biological warfare. In either case, we are all in this process together, and how it evolves and how we adapt is entirely in our hands. The decisions we make now will not only determine our viability as a species in the near future but will have ripple effects throughout many generations into the future, so it is absolutely critical we get this right, and we do it in a way that does not reinforce species dominance — or what I would also like to coin “species virulence”.
If you are dealing with challenges in physical health or cognitive ability and need additional insights into how to develop aggressive and effective strategies for immune resilience / protection, I encourage you to reach out today and book a consultation with Transcend Genomics. Your needs, as an individual, cannot be determined by broad, generalized studies that draw conclusions based on limited evidence. Everyone needs their own, specific approach, tailored to their history, current health challenges, genetics, and all other vital markers that define how you function biochemically.
Ultimately, there are no silver bullets for COVID-19. As much as the public would like to believe that a vaccine or an antibody-based strategy — or even ascorbic acid or melatonin — could in any way have profound impacts on the outcomes of this pandemic, there is still a profound need to think outside of the box and realize strategies that not only carry us through to the other end of this current viral cycle, but also ensure our ability to both thrive and enjoy well-being without sacrificing our own health and the health of our environment.
Join our program today!
Transcend Genomics offers an unparalleled opportunity for self-exploration, growth, and radical body-mind optimization. The journey begins with an analysis of over 20,000 genes and 200 metabolic pathways to discover the areas of your genome that could have the greatest impact on your health, cognition, and longevity.